Глава 5 ЗАБОЛЕВАНИЯ СИСТЕМЫ КРОВИ
5.1. АНЕМИИ
Анемиями называются такие заболевания, при которых количество гемоглобина и/или эритроцитов крови не достигает установленных для данной возрастной группы физиологических величин. На их долю приходится от 8 до 12 случаев на 100 тыс. населения, причем женщины болеют чаще мужчин (1,8: 1,0).
Основные причины возникновения анемий: а) недостаточное образование в костном мозге гемоглобина и/или эритроцитов; б) повышенное разрушение эритроцитов; в) кровопотери.
Условно все многообразие анемий можно разделить на две большие группы: 1) гипорегенераторные (с уменьшенной продукцией костным мозгом эритроцитов и/или гемоглобина); 2) гиперрегенераторные. К первым относят апластические анемии, В12-, фолиеводефицитные, железодефицитные, сидеробластные и талассемии, ко вторым – гемолитические и постгеморрагические. Общепринятой классификации анемий нет, поскольку в большинстве случаев малокровие – симптом ряда заболеваний, а не нозологическая форма. Приводим одну из наиболее распространенных классификаций анемий.
Классификация анемий (по: P. R. Reich, 1978).
I. Морфологическая:
– нормоцитарная нормохромная (кровопотери, гемолитические, апластические);
– микроцитарная гипохромная (железодефицитная, сидеробластная, отравление свинцом);
– макроцитарная гипер- и нормохромная (мегалобластные, болезни печени, предлейкозы).
II. Физиологическая:
– гипорегенераторные (апластическая анемия, токсины, хронические заболевания, дефицит железа);
– с повышенным разрушением или потерей эритроцитов (гемолитические анемии, кровопотери);
– с нарушением созревания эритроцитов (мегалобластные анемии, предлейкозы, сидеробластная анемия, талассемии).
III. Клиническая:
– гипохромные (железодефицитная, сидеробластная);
– мегалобластные (В12-дефицитная, фолиеводефицитная);
– гемолитические (Кумбс-позитивная, Кумбс-негативная);
– апластические (связанная с приемом лекарств, идиопатическая);
– постгеморрагические (острые и хронические кровопотери);
– миелофтиз (лейкемии, лимфомы, миелофиброз).
Клинические проявления анемии различны. Их появление во многом связано со степенью и темпом снижения уровня гемоглобина и/или эритроцитов и, отсюда, с выраженностью компенсаторной реакции дыхательной и сердечно-сосудистой систем больного на анемию. В итоге у части больных жалоб может не быть. В других ситуациях беспокоят одышка инспираторного типа и сердцебиение. Может быть слабость разной степени выраженности, головокружения, шум в голове. Объективно у больных обнаруживают бледность кожных покровов и слизистых, умеренное понижение артериального давления с появлением систолического шума на верхушке сердца и на сосудах. Кроме того, в клинической картине анемии могут быть представлены общие симптомы дефицита железа, недостатка витамина В12 или фолиевой кислоты, некоторые отклонения в обмене гема, глобина и т. д.
5.1.1. Острая постгеморрагическая анемия
Определение. Острой постгеморрагической анемией называют анемию, которая развивается после потери значительного количества крови.
Патогенез острой постгеморрагической анемии связан с резким уменьшением объема циркулирующей крови, особенно ее плазменного компонента. В первую очередь это приводит к развитию острой гипоксии и связанных с ней одышке и сердцебиению. В связи с гипоксией повышается содержание эритропоэтина в сыворотке крови, что приводит к активации эритроидного ростка костного мозга и к увеличению в крови содержания ретикулоцитов.
Клиническая картина этого типа анемии зависит от объема потерянной крови, скорости ее истечения, а также источника кровотечения. При больших кровотечениях на первое место в клинике выходит картина коллапса. Это проявляется резкой слабостью, головокружением, бледностью кожных покровов и слизистых, сухостью во рту, холодным потом и рвотой. Регистрируется снижение артериального и венозного давления. Уменьшается сердечный выброс крови и учащается пульс, который может стать слабым и даже нитевидным.
Лабораторная диагностика. Содержание гемоглобина и эритроцитов в крови в момент кровотечения может быть не изменено. Характерно повышение ретикулоцитов крови как адаптационная реакция на кровотечение, а также разной степени выраженности нейтрофильный лейкоцитоз. В случае же присоединения анемии, которая развивается у больных с кровотечением не сразу, а на второй-третий день после кровотечения, когда с заместительной целью в кровь поступает тканевая жидкость, она носит нормоцитарный и нормохромный характер.
Диагноз скрытого от глаз врача массивного кровотечения прежде всего основывается на перечисленных выше клинических признаках, которые могут быть подкреплены обнаружением в крови повышенного количества ретикулоцитов, пробой Грегерсена и т. д.
Пример формулировки диагноза:
Язвенная болезнь желудка. Желудочное кровотечение. Острая постгеморрагическая анемия.
Дифференциальный диагноз. При отсутствии явных признаков кровотечения дифференциальный диагноз острой постгеморрагической анемии следует проводить с остро протекающим гемолитическим кризом. Правильный диагноз помогает поставить наличие в анамнезе больных гемолитической анемией повторяющихся желтух или темной мочи, других клинических признаков гемолиза, обнаружение в крови повышенного количества непрямого билирубина, снижение гаптоглобина сыворотки крови, положительная проба Кумбса и т. д., а при объективном исследовании – увеличение размеров селезенки.
Лечение острой постгеморрагической анемии начинают с остановки кровотечения и с проведения противошоковых мероприятий.
Показаниями к началу трансфузионной терапии являются: а) продолжающееся кровотечение; б) существенное падение систолического артериального давления (ниже 90 мм рт. ст.); в) учащение пульса на 20 и более ударов в минуту. При кровопотере от 500 мл крови до 1 л она может быть восполнена плазмозамещающими растворами, в том числе полиглюкином, желатинолем, альбумином, физиологическим раствором, раствором Рингера и т. д. В случае же большей кровопотери оправданно введение донорской крови, хранящейся не более 5 дней. Следует помнить, что до введения эритроцитарной массы ее желательно развести полиглюкином в отношении 1: 2. Кроме того, при массивной кровопотере большое значение имеет скорость введения трансфузионных сред. При этом из-за резкого снижения венозного давления и спадения локтевых вен можно пользоваться пункцией подключичных вен или венесекциями с последующей одновременной струйной трансфузией в 2 – 3 вены. Наконец, во избежание развития «синдрома массивных трансфузий» недопустимо восполнение всей кровопотери кровью.
5.1.2. Гипохромные анемии
Характерным признаком этой патогенетически разнородной группы заболеваний является гипохромия, сочетающаяся с уменьшением размера эритроцитов. В основе развития гипохромных анемий может быть: а) дефицит железа; б) нарушение синтеза порфиринов; в) нарушение синтеза цепей глобина.
Дефицит железа в организме возникает в результате: а) кровопотерь; б) повышенного его потребления, например у беременных женщин; в) нарушения всасывания; г) нарушения транспорта в крови.
Патогенез. Поскольку железо является одним из самых распространенных элементов земной коры, оно играет важную роль в организме человека. Большая часть его (3 из 5 г) приходится на гемоглобин эритроцитов. Меньшая, приблизительно 0,6 г, входит в состав миоглобина и дыхательных ферментов, а около 1,5 г находится в депо в виде ферритина и гемосидерина. Суточные потери железа составляют всего 2 мг и обусловлены выделением с желчью, мочой и потом, а также в составе слущивающегося эпителия. Восстановление утраченного железа происходит за счет всасывания пищевого железа в двенадцатиперстной кишке и меньше – в тощей кишке. Обычно за сутки может всосаться не более 3,5 мг железа, хотя в условиях дефицита доля абсорбируемого железа увеличивается в 2 – 3 раза. Контроль за поступлением в портальный кровоток железа осуществляется энтероцитами, которые могут связать избыток железа с ферритином и возвратить его в просвет кишки. Из кишечной стенки железо в виде Fe3+ поступает в плазму в комплексе с трансферрином, каждая молекула которого может связать два атома железа. Благодаря наличию на эритроидных элементах костного мозга специальных рецепторов к трансферрину, последний передает принесенное им железо по прямому назначению, а после освобождения готов к его транспорту вновь (см. цв. вкл., рис. 5.1). У здоровых лиц железом насыщена только треть трансферрина. Эта пропорция в течение суток может широко варьировать главным образом из-за меняющихся способностей самих макрофагов отдавать железо трансферрину.
После разрушения эритроцитов железо не теряется из организма, а накапливается в окисленном состоянии (Fe3+) в макрофагах ретикулоэндотелиальной системы в составе ферритина и/или гемосидерина. Ферритин представляет собой водорастворимое соединение с молекулярным весом 465 000. Оно состоит из наружной белковой оболочки – апоферрина и железа, на долю которого приходится до 20 % массы ферритина. Что касается гемосидерина, доля железа в котором доходит до 37 %, то он представляет собой частично переваренные в лизосомах агрегаты ферритина. Железо восстанавливается до Fе2+ при участии аскорбиновой кислоты и в таком виде мобилизуется для нужд организма. Однако для переноса трансферрином оно вновь окисляется церулоплазмином до Fе3+. Учитывая то, что суточная потребность в железе, составляющая 5 мг, содержится в 10 мл крови, при кровопотерях, так же как при повышенных запросах, запас железа в организме быстро истощается.
Клиническая картина. У одних больных железодефицитными анемиями (ЖДА) синдром сидеропении выражен незначительно, а у других обнаруживает себя еще до снижения гемоглобина. Из-за уменьшения миоглобина в мышцах больных может беспокоить выраженная мышечная слабость (до дисфагии и недержания мочи). Характерны сухость и трещины кожи на руках и ногах, глоссит и ангулярный стоматит (заеды в углах рта), уплощенные, тонкие, исчерченные, легко ломающиеся ногти (койлонихии), тонкие, легко выпадающие волосы, ахлоргидрия. Последней сопутствует снижение аппетита, чувство тяжести в эпигастральной области, извращение вкуса и обоняния. В частности, больные с железодефицитными анемиями любят есть мел, глину, известь. Им нравится запах бензина, керосина, гуталина и т. д.
На фоне выраженного падения гемоглобина до 70 г/л и более, помимо признаков сидеропении (или без них), могут появиться выраженное головокружение, головные боли, а также сердцебиение и одышка как компенсаторные реакции сосудистой и дыхательной систем на гипоксию.
При объективном обследовании у этих больных может быть обнаружена бледность кожных покровов и слизистых, одышка инспираторного характера, тахикардия, систолический шум на верхушке и на сосудах сердца.
Лабораторная и инструментальная диагностика. При наличии у больного анемией клинических признаков сидеропении диагноз ЖДА не труден. В отсутствие таковых следует ориентироваться не только на низкий цветовой показатель, но и на снижение в сыворотке крови железа и ферритина, а также повышение ненасыщенного сидерофилина. Содержание ретикулоцитов в крови нормальное или умеренно сниженное. Оно может увеличиться только после острой кровопотери. Лейкоцитарная формула практически не изменена. Содержание тромбоцитов имеет тенденцию к повышению. Пунктат костного мозга богат клеточными элементами с гиперплазией эритроидного ростка. Количество сидеробластов в костном мозге снижено (< 15 %).
Уточнение причины железодефицитной анемии направлено на поиск кровотечений, прежде всего генитальных и из желудочно-кишечного тракта. Это может быть гиперполименоррея, а также язвенная болезнь, болезнь Крона, рак, дивертикулы желудка, диафрагмальная грыжа, геморрой, портальная гипертензия и т. д. Для исключения кровотечений из желудочно-кишечного тракта необходима постановка реакции Грегерсена, а в случае ее нечувствительности даже проба с меченными Cr51 эритроцитами. Показана консультация гинеколога, фиброгастроскопия, колоноскопия, стандартное и нестандартное, например, с двойным контрастированием, рентгенологическое исследование желудочно-кишечного тракта, а при необходимости даже выполнение диагностической лапаротомии. В случае неуспеха выявления источника кровотечения у больных с немотивированной железодефицитной анемией следует иметь в виду, что у части больных такие кровотечения могут происходить в замкнутые пространства, откуда железо извлекается организмом плохо. Речь идет о так называемых гломусных опухолях желудка и кишечника, миомах матки, а также первичном легочном сидерозе. Кроме того, несомненные трудности для возвращения в кроветворение захваченного макрофагами железа встречаются у больных с хроническими воспалительными заболеваниями.
Диагноз ЖДА формулируется с указанием этиологии заболевания.
Пример формулировки диагноза:
Железодефицитная анемия, повторные геморроидальные кровотечения.
Дифференциальный диагноз железодефицитной анемии следует проводить с другими видами анемий, и прежде всего с гипохромными. Как и при постановке диагноза, решающим моментом для разграничения этих заболеваний может быть наличие в клинике симптомов сидеропении. При отсутствии таковых обращают внимание на низкий уровень железа и ферритина сыворотки, высокий свободный сидерофилин, уменьшение в костном мозге содержания сидеробластов (< 15 %). В трудных случаях для подтверждения в организме скрытого дефицита железа может быть проведен десфераловый тест, основанный на уменьшенном выведении с мочой больными ЖДА связываемых десфералом солей железа.
Лечение больных ЖДА может проводиться в амбулаторных условиях. Терапией выбора являются препараты для перорального применения. В настоящее время их предложено довольно много. Наиболее эффективные: ферроплекс, конферон, феррокаль, орферон, ферроградумет, фенюльс, гемофер пролонгатум, сорбифер дурулес, тардиферон, апо-ферроглюконат, железа фумарат, мальтофер, феррум лек, венофер и др. Они принимаются сразу после приема пищи из расчета 5 мг/кг массы тела в день. Для большинства препаратов непролонгированного действия максимальная доза препарата 6 таблеток в сутки. В то же время для таких пролонгированных препаратов, как ферроградумет и фенюльс, достаточно и одной таблетки в сутки. В качестве усиливающего всасывание железа средства может быть использована аскорбиновая кислота в дозе 200 мг на 1 г железа.
На фоне приема препаратов железа самочувствие больного быстро улучшается, несмотря на то что уровень гемоглобина поднимается довольно медленно.
Этот парадоксальный эффект связан с общим укрепляющим действием железа на организм, в том числе в плане повышения миоглобина, цитохромов и т. д. Лечение больного с доказанной железодефицитной анемией должно проводиться непрерывно в течение 3 – 4 мес. и быть направлено не только на нормализацию гемоглобина, но и устранение всех симптомов сидеропении и создание депо железа. При отсутствии эффекта от пероральных препаратов показаны парентеральные (феррум лек 100 мг внутримышечно, мальтофер, венофер и т. д.). Однако из-за возможной индивидуальной непереносимости вводить их следует осторожно, а начинать лечение с половинной дозы.
Профилактический прием препаратов железа необходим донорам, беременным, кормящим, а также обильно менструирующим женщинам. В последнем случае желателен ежемесячный (2 – 4 дня) прием препаратов железа внутрь.
Второй вид гипохромной анемии – так называемая сидеробластная, или сидероахрестическая анемия. Она связана с нарушением синтеза порфиринов и характеризуется наличием в крови гипохромных микроцитарных эритроцитов, а в костном мозге – кольцевидных сидеробластов. Встречается это заболевание довольно редко. В части наблюдений оно имеет отчетливую наследственную природу, в части носит приобретенный характер.
Этиология. Наследование врожденной формы сидеробластной анемии связано с X-хромосомой и поэтому чаще наблюдается у мужчин. Выделяют также специальный «пиридоксинчувствительный» вариант врожденной сидеробластной анемии, при котором может быть достигнута частичная гематологическая ремиссия на фоне высоких доз витамина В6 (2 – 4 мг/кг/сут).
Приобретенная сидеробластная анемия обычно возникает без отчетливой связи с внешними факторами или заболеваниями. У части больных сидеробластную анемию могут провоцировать: свинцовая интоксикация, длительный прием алкоголя, лечение изониазидом и левомицетином. Кроме того, идиопатическая приобретенная сидеробластная анемия может быть предстадией лейкоза, о чем пойдет речь в разделе «Миелодиспластические синдромы».
Патогенез сидеробластной анемии связан с дефектом синтеза гема в митохондриях эритроидных клеток. Речь идет о нарушении функции синтетазы дельта-аминолевулиновой кислоты и ряда других митохондриальных ферментов. Основным предшественником дельта-аминолевулиновой кислоты в клетке являются сукцинил-коэнзим А и глицин. Ее синтез стимулируется эритропоэтином, а катализируется производным витамина B6 – пиридоксаль-5-фосфатом. Образующийся при этом из дельта-аминолевулиновой кислоты протопорфирин в ходе дальнейших превращений соединяется с двухвалентным железом (Fe2+) и формирует гем. В свою очередь, каждая молекула гема на полирибосомах связывается с глобиновой цепью. Комбинация же четырех цепей глобина с гемом и представляет собой наш гемоглобин.
Если представить теперь, что образование дельта-аминолевулиновой кислоты и протопорфиринов в эритроидных клетках будет нарушено, это приведет, с одной стороны, к накоплению в митохондриях двухвалентного железа, с другой – к недостаточному синтезу гемоглобина. В итоге разовьется анемия, при которой будут иметь место и гиперсидеринемия, и резкое увеличение в костном мозге содержания кольцевидных сидеробластов (нормобластов с расположенными вокруг ядра – в митохондриях – гранулами железа), которые могут быть выявлены специальными красителями. В зависимости от характера анемии (врожденная или приобретенная), а также от непосредственной ее причины механизмы повреждения синтеза гема могут различаться. Так, в случае свинцовой интоксикации наряду с нарушением синтеза гема и глобина происходит отчетливое торможение работы фермента пиримидин-5S-нуклеотидазы, что, с одной стороны, приводит к кумуляции в эритроцитах денатурированной РНК, с другой – к появлению в них базофильной пунктации.
Клиническая картина заболевания проявляет себя анемическим синдромом разной степени тяжести. Некоторые больные нуждаются в трансфузиях эритроцитарной массы. Вследствие неэффективного эритропоэза и внутрикостномозгового гемолиза у них может иметь место некоторая желтушность кожи и склер. Кроме того, нередко определяются умеренная спленомегалия и гепатомегалия. Наконец, у больных с врожденным вариантом могут быть зафиксированы такие признаки гемохроматоза, как гиперпигментация кожи, диабет, дисфункция печени и сердца и т. д. В случае со свинцовой интоксикацией на первое место в клинике могут выступать такие ее яркие проявления, как сильные схваткообразные боли в животе, явления полиневрита, изменение со стороны слизистой полости рта (свинцовая кайма на деснах).
Диагноз сидеробластной анемии должен обсуждаться врачами в случае хорошо верифицированной гипохромной анемии без клинических проявлений сидеропении и при наличии в сыворотке крови высокого содержания железа и ферритина, а также низкого содержания ненасыщенного сидерофилина. Подтверждается диагноз исследованием пунктата костного мозга, выявляющего большое количество кольцевидных сидеробластов. При этом может быть заказан цитoгeнeтичecкий анализ клеток костного мозга, который в случае выявления клоновых изменений кариотипа позволит уверенно диагностировать один из трудно определяемых обычными способами подвариантов миелодиспластического синдрома, а именно рефрактерную сидеробластную анемию, и выбрать правильную тактику ее ведения.
Дифференциальный диагноз сидеробластной анемии проводится с другими видами анемий, и прежде всего с гипохромными. В отличие от железодефицитной анемии, при этой патологии в клинике не будут представлены симптомы сидеропении, а в лабораторных анализах уровень железа не только не снижен, а даже повышен. В отличие от талассемии, дефекты костной системы, так же как спленомегалия, для сидеробластной анемии не характерны. В трудных случаях решающим в распознавании сидеробластной анемии от других анемий может быть высокое содержание в костном мозге кольцевидных сидеробластов, а в ряде случаев также положительный эффект терапии большими дозами витамина В6.
Лечение сидеробластной анемии вызывает у врача много проблем. При наличии выраженной анемии показана заместительная терапия эритроцитарной массой, дополняемая введением десферала для профилактики гемосидероза. При исключении миелодиспластического синдрома оправданна также длительная (до 2 мес.) терапия большими дозами (2 – 4 мг/кг/сут) витамина В6. К сожалению, утешительного эффекта данной терапии удается достичь только у единичных больных с врожденной пиридоксин-чувствительной сидеробластной анемией.
Если такая терапия неэффективна, могут быть предложены для лечения большие дозы андрогенов.
Для профилактики развития гемохроматоза и его осложнений при высоких показателях ферритина можно использовать хелаторы железа (десферал или эксиджад). Описаны также единичные случаи аллогенной трансплантации гемопоэтических стволовых клеток для лечения врожденных форм сидеробластной анемии.
Прогноз заболевания остается крайне серьезным. Основными причинами смерти больных являются осложнения со стороны сердечно-сосудистой и эндокринной систем, печени и почек, в том числе спровоцированные гемохроматозом.
Другим видом гипохромных анемий являются талассемии, которые представляют собой сборную группу заболеваний наследственной природы, при которых нарушен синтез одной или более структурно неизмененных цепей глобина. Количество последних у человека достигает четырех. Отсюда возможны четыре вида талассемии, из которых больший клинический интерес представляют áá-талассемия и â-талассемия. Первая больше распространена в Китае, Индии и на Дальнем Востоке, вторая – в Средиземноморье, Малой и Средней Азии.
á-Талассемия. Подавляющее большинство á-талассемий связано с делецией генов á-цепей глобина, находящихся на хромосоме 16, а клинические и гематологические проявления зависят от количества пораженных генов. При вовлечении одного из четырех генов á-цепей глобина (á-/áá) их носительство не сопровождается ни клиническими, ни гематологическими изменениями. При поражении двух из четырех генов á-цепей глобина (á-/á— или – /áá) определяется микроцитоз и гипохромия, хотя выраженной анемии нет. Делеция трех генов áá-цепей глобина (-/á-) сопровождается развитием гипохромной микроцитарной анемии с мишеневидными эритроцитами (см. цв. вкл., рис. 5.2), тельцами Гейнца в них и умеренным гемолизом. Несмотря на такие морфологические изменения эритроцитов, тяжелого анемического синдрома не развивается, что чаще всего позволяет обходиться даже без гемотрансфузий. Полное отсутствие генов á-цепей несовместимо с жизнью.
â-Талассемия. В отличие от двух генов á-цепи глобина человек наследует от родителей только по одному гену â-цепи, которые расположены на хромосоме 11. Отсюда возможны гетерозиготные, гомозиготные и двойные гетерозиготные варианты талассемии с уменьшением в крови нормального, состоящего из двух áá— и двух â-цепей, гемоглобина Нb А и увеличением гемоглобинов Нb F и Нb А2.
Патогенез. Молекулярные механизмы патогенеза â-талассемии сложнее и разнообразнее, чем при á-талассемии. Так, делеции генов при этом виде талассемии не бывает. Вместо этого выявляются точечные мутации, которых на сегодняшний день описано более 100. В результате мутаций при гетерозиготной талассемии возникает дисбаланс синтеза á-иâ-цепей глобина; развивается неэффективный эритропоэз и гемолиз различной степени тяжести. При гомозиготной ââ-талассемии эти процессы выражены значительно сильнее и проявляют себя как костномозговым разрушением эритроидных элементов, так и массивным повреждением эритроцитов в селезенке. Как следствие этого, развиваются экстрамедуллярные очаги компенсаторного кроветворения.
Клиническая картина талассемии разнообразна. Для «большой» гомозиготной â-талассемии характерен выраженный анемический синдром, который выявляется очень рано (в возрасте 3 – 6 мес.). Из-за гемолиза эти дети отстают в развитии, имеют место деформации костей черепа, истончение коркового вещества костей, спонтанные переломы. Характерна окраска кожи, обусловленная бледностью, желтухой и отложением меланина. Резко увеличены размеры селезенки и печени. Выявляется кардиомегалия и признаки сердечной недостаточности. Могут быть клинические проявления гемохроматоза.
Клинические проявления гетерозиготной â-талассемии менее ярки. В большинстве случаев заболевание выявляется случайно. Оно проявляет себя умеренно выраженным анемическим синдромом, реже небольшой спленомегалией. Желтуха встречается редко.
Лабораторная диагностика талассемии зависит от варианта заболевания. У больных гетерозиготной á-талассемией выявляется разной степени выраженности гипохромная микроцитарная анемия. Встречаются отдельные мишеневидные эритроциты, а также клетки с базофильной пунктацией. Содержание ретикулоцитов и сывороточного железа обычно нормальное. Электрофорез гемоглобина выявляет двухкратное увеличение количества гемоглобина А2, которого при áá-талассемии и других гипохромных анемиях нет. У половины больных может быть также повышен фетальный Нb F.
Приблизительно такие же лабораторные признаки находят при малой á-талассемии. Однако электрофорез гемоглобина патологии не выявляет. Для á-талассемии с поражением 3 генов цепей глобина (болезнь Нb Н) характерно наличие в клетках гемоглобина Н на фоне снижения количества гемоглобина А. Для гомозиготной â-талассемии характерны выраженная гипохромная анемия, увеличение содержания ретикулоцитов в крови, нормоцитоз, мишеневидные эритроциты и их базофильная пунктация. При электрофорезе гемоглобина выявляется резкое снижение или отсутствие гемоглобина А при значительном увеличении гемоглобина F. Кроме того, может быть умеренно увеличено количество гемоглобина А2.
Дифференциальный диагноз талассемии проводится с другими гемолитическими и гипохромными анемиями. В отличие от ЖДА, у больных талассемией отсутствуют синдромы сидеропении, не снижено содержание железа и ферритина в сыворотке крови и, наоборот, не наблюдается увеличение уровня ненасыщенного сидерофилина. В отличие от других гемолитических анемий, при талассемиях, за редким исключением, имеют место гипохромные микроцитарные эритроциты. В трудных случаях помогает электрофорез гемоглобинов, выявляющий их аномальные формы.
Лечение. Больные малыми талассемиями в специальной терапии не нуждаются. Лечение больших талассемий включает: трансфузии эритроцитов после индивидуального подбора доноров по системе HLA, прием фолиевой кислоты (достаточно 5 мг/сут), введение десферала внутривенно 1 – 2 г на каждые 500 мл донорской крови и аскорбиновой кислоты (200 мг/сут), спленэктомию, лечение органных поражений, связанных с гемосидерозом. При наличии HLA-совместимых доноров костного мозга молодым больным (до 16 лет) может быть проведена аллогенная трансплантация, после которой безрецидивная выживаемость достигает 80 %.
Прогноз для больных малыми формами талассемии благоприятен, а для гомозиготной â-талассемии серьезен. Ее основными осложнениями, кроме замедленного развития ребенка, являются инфекции, в том числе вирусные гепатитыВиС,игемосидероз внутренних органов, связанный с множественными гемотрансфузиями.
5.2. АНЕМИИ ПРИ ХРОНИЧЕСКИХ ЗАБОЛЕВАНИЯХ
Определение. Анемию, ассоциированную с инфекционными и воспалительными процессами и онкологией, называют анемией хронического заболевания или анемией на фоне хронического заболевания. Поначалу казалось, что они связаны с нарушенным усвоением железа участвующими в воспалении макрофагами. Однако позднее проявился мультифакториальный генез анемии хронического воспаления, при которых помимо нарушения усвоения железа были еще представлены: а) укорочение длительности жизни эритроцитов; б) снижение в крови уровня эритропоэтина.
Патогенез. Создается впечатление, что ведущую роль в патогенезе этих анемий играет гиперпродукция различных цитокинов, прежде всего фактора некроза опухоли, интерлейкинов 1 и 6, а также интерферонов á, â и ã. В основе развития анемического синдрома при анемии хронического заболевания лежит уменьшение длительности жизни эритроцитов и недостаточная компенсаторная реакция эритропоэза, что обусловлено ингибирующим влиянием цитокинов на эритропоэз и чувствительностью эритроидных клеток-предшественников к эритропоэтину. Усвоение же железа эритроидными клетками-предшественниками может быть не изменено или нарушено, что может являться дополнительным фактором развития анемии. Отсюда понятно, что из этой группы должны быть исключены анемии, обусловленные замещением костного мозга опухолевыми клетками, потерей крови, заболеваниями печени, почек, почечной недостаточностью и эндокринопатиями, хотя многие из этих заболеваний по своей сути тоже хронические.
Распространенность анемий на фоне хронического заболевания. Учитывая большую распространенность среди населения хронических заболеваний воспалительного или опухолевого генеза, эти виды анемии встречаются довольно часто. Основными их причинами являются: а) все хронические инфекции; б) хронические неинфекционные воспалительные заболевания, в том числе ревматоидный артрит, ревматическая лихорадка, системная красная волчанка, тяжелая травма, ожоги, васкулиты и др.; в) злокачественные опухоли и гемобластозы; г) алкогольное поражение печени; д) тяжелая сердечная недостаточность; и др.
Клинические проявления у больных анемией и ее тяжесть в подавляющем числе наблюдений зависят от тяжести основной патологии. Чаще всего на ранних стадиях заболевания имеет место легкая, реже – средней тяжести анемия с колебаниями гематокрита от 0,25 до 0,4, что напрямую зависит от колебаний объема циркулирующей крови и объема циркулирующей плазмы. При высоком уровне в крови интерлейкина 6 на анемию хронического заболевания может наслаиваться «анемия разведения».
Лабораторная диагностика. Для анемии хронического заболевания типично нормальное или сниженное содержание ретикулоцитов в крови. Анемия обычно носит нормоцитарный и нормохромный характер, а в 23 – 50 % случаев может быть микроцитарной и гипохромной. Последний симптом встречается чаще, чем микроцитоз эритроцитов, и нередко может быть одним из первых признаков анемии хронического заболевания.
Также для анемии хронического заболевания характерно снижение показателя сывороточного железа в отсутствие увеличения или даже при наличии снижения общей железосвязывающей способности сыворотки. При этом показатель насыщения трансферрина чаще находится на субнормальном уровне. Снижение сывороточного железа отмечается в ранние сроки тяжелого инфекционного процесса и опережает развитие анемии. В свою очередь, это сопровождается умеренным снижением в костном мозге числа сидеробластов (до 5 – 15 %) и увеличением гемосидерина в макрофагах. Уровень ферритина, как правило, повышен, хотя при сопутствующей ЖДА, которая встречается в 20 % наблюдений, может стать относительно низким.
Важным показателем анемии хронического заболевания является увеличение содержания в крови таких острофазовых белков, как фибриноген, церулоплазмин, гаптоглобин, СРБ, С3-компонент комплемента и амилоид А протеин. Часто у больных анемией хронического заболевания имеет место увеличение туморнекротического фактора, интерлейкинов 1 и/или 6 и интерферонов (прежде всего â-иã-интерферонов). В то же время уровни альбумина и трансферрина, как правило, снижены.
Диагноз анемии на фоне хронического воспаления ставится по данным отмеченного выше нарушения обмена железа, сочетающегося с одной или несколькими из перечисленных выше патологий.
Дифференциальный диагноз, прежде всего, проводится с железодефицитными анемиями. В отличие от ЖДА, при анемии хронического заболевания на фоне снижения содержания железа в сыворотке отсутствует снижение запасов железа в ретикулоэндотелиальной системе и сидеропенический синдром. Кроме того, уровень ферритина в сыворотке крови при этих видах анемий, как правило, повышен.
Лечение. Основой терапии анемии, обусловленной хроническими воспалительными заболеваниями, является лечение основной патологии. В том случае, если из-за анемического синдрома тяжесть основного патологического процесса усугубляется, т. е. усиливается гипоксемия, значимо снижается качество жизни, есть все основания для проведения терапии рекомбинантным эритропоэтином. Стандартная схема лечения подразумевает применение эритропоэтина (эпрекс, рекормон и др.) по 30 МЕ 3 раза в неделю. При этом повышение гемоглобина может быть зафиксировано уже через 3 – 4 нед. Если же эффекта от применения эритропоэтина нет или снижение гемоглобина достигает критических цифр (< 50 г/л), то по жизненным показаниям следует применять трансфузии эритроцитарной массы.
5.3. МАКРОЦИТАРНЫЕ (МЕГАЛОБЛАСТНЫЕ) АНЕМИИ
Мегалобластные анемии представляют собой группу заболеваний, для которых характерны связанные с нарушением синтеза ДНК мегалобластное кроветворение и макроцитоз крови. В основе заболеваний лежит нарушение естественных путей синтеза пуринов и пиримидинов и торможение ДНК-полимеризации. Основными причинами развития макроцитарных анемий считаются дефицит фолиевой кислоты и/или витамина В12.
Этиология и патогенез. Роль витамина В12 и фолиевой кислоты показана на схеме 5.1, из которой видно, что недостаток фолиевой кислоты, и в частности 5, 10-метил-тетрагидрофолат-полиглутамата, приводит к торможению превращения дезоксиуридин монофосфата в дезокситимидин монофосфат и в результате – к торможению синтеза ДНК. В то же время витамин В12 прямого участия в синтезе ДНК не принимает. Однако он необходим клетке для превращения поступающего из кишечника 5-метил-тетрагидрофолата (фолиевой кислоты) в активные коэнзимные формы, в том числе упомянутого выше полиглутамата.
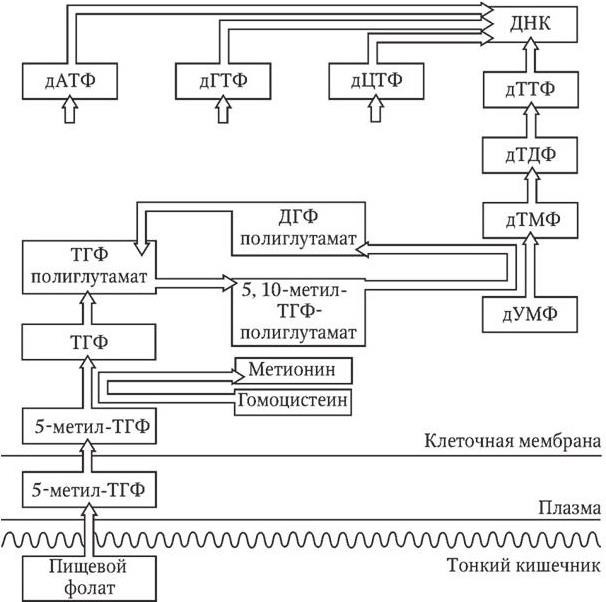
Схема 5.1. Место фолиевой кислоты и витамина В12 в синтезе ДНК, связанное с превращением дезоксиуридинмонофосфата (дУМФ) в дезокситимидинмонофосфат (дТМФ) и далее дезокситимидиндифосфат (дТДФ), дезокситимидинтрифосфат (дТТФ) и тимидин:
ДГФ – дигидрофолат; ТГФ – тетрагидрофолат; дАТФ – дезоксиаденозинтрифосфат; дГТФ – дезоксигуанитидинтрифосфат; дЦТФ – дезоксицитидинтрифосфат
Наиболее частой причиной дефицита фолиевой кислоты является неадекватное поступление ее с пищей, в первую очередь на фоне повышенной потребности. Последняя имеет место у беременных женщин и кормящих матерей, у быстрорастущих детей, особенно в условиях искусственного вскармливания, при гемолизе, некоторых видах злокачественных и воспалительных заболеваний (эксфолиативный дерматит, псориаз, болезнь Крона и т. д.). Другой причиной дефицита фолиевой кислоты может быть нарушение ее всасывания в кишечнике (болезнь Крона, спру) и плохое усвоение. Последнее может иметь место при длительном приеме некоторых противосудорожных (дифенин, барбитураты), противотуберкулезных (циклосерин), противозачаточных и противодиабетических (метформин) препаратов и алкоголя. Наконец, дефицит фолиевой кислоты может возникнуть в результате приема больным прямого ингибитора дигидрофолатредуктазы – метотрексата.
Основной причиной дефицита в организме витамина В12 следует считать нарушение его всасывания в кишечнике из-за недостаточной выработки желудком фактора Кастла. Последнее имеет место у больных атрофическим гастритом, с опухолью желудка, а также может быть следствием ранее перенесенных гастрэктомий и серьезных заболеваний тонкого кишечника. Другой причиной дефицита витамина В12 может быть носительство широкого лентеца, который поглощает этот витамин в большом количестве. Значительно реже дефицит витамина В12 возникает из-за недостатка его в пище, например, у строгих вегетарианцев. Кроме того, нарушение всасывания и усвоения витамина В12, как и фолиевой кислоты, может наблюдаться на фоне приема противотуберкулезных (ПАСК) и противодиабетических (метформин) препаратов, некоторых антибиотиков (неомицин) и алкоголя. Кратковременное снижение активности витамина В12 можно наблюдать в случае применения анестезии закисью азота. Наконец, у новорожденных мегалобластная анемия может возникнуть из-за дефицита в сыворотке транскобаломина II.
Хорошо известно, что основными источниками фолиевой кислоты являются листья растений, а витамина B12 – продукты животного происхождения (мясо, печень, рыба). Вместе с тем его довольно много в сое и некоторых морских водорослях (хиджики, морская капуста). Наконец, он может синтезироваться многими микроорганизмами, в том числе населяющими наш кишечник. Запасов витамина B12 в организме обычно хватает на 5 – 6 лет, а фолиевой кислоты – на несколько месяцев. В желудке витамин В12 высвобождается из белковых комплексов. Далее он связывается с упомянутым выше гликопротеином – внутренним фактором Кастла, который синтезируется париетальными клетками слизистой оболочки желудка. Этот комплекс «витамин/фактор Кастла» соединяется со специфическими рецепторами в дистальных отделах подвздошной кишки, откуда витамин В12 поступает в портальный кровоток в связи с его основным переносчиком – транскобаламином II. При этом внутренний фактор в кровоток не поступает.
Клиническая картина. Мегалобластная В12-дефицитная анемия встречается чаще у женщин, чем у мужчин (1,6: 1,0). Средний возраст больных – старше 60 лет. Поскольку развитие основной причины В12-дефицитной анемии – атрофического гастрита – носит аутоиммунный характер, это заболевание часто сочетается у больных с аутоиммунным тиреоидитом, болезнью Грейвса, иммунным гипопаратиреозом, витилиго, болезнью Адиссона и гипогаммаглобулинемией. Замечено также, что эта анемия может быть уделом лиц, имеющих группу крови II (А), голубые глаза, и обычно сопровождается ранним поседением. У большинства больных В12-дефицитными анемиями (до 90 %) могут быть обнаружены антитела к париетальным клеткам слизистой желудка, у половины – к внутреннему фактору Кастла. С одной стороны, они могут препятствовать связыванию витамина B12 с внутренним фактором, с другой – препятствовать его всасыванию в подвздошной кишке. Следует также заметить, что антитела к внутреннему фактору Кастла для B12-дефицитных анемий являются специфическими, в то время как антитела к париетальным клеткам желудка у части пожилых женщин могут быть обнаружены и в отсутствие анемии.
Клиническая симптоматика у больных В12-дефицитной анемией развивается медленно. Для нее характерен анемический синдром, симптомы поражения желудочно-кишечного тракта и, реже, нервной системы в виде так называемого фуникулярного миелоза. По описанию И. А. Кассирского, клиническая картина синдрома складывается из сочетаний спастического спинального паралича и табетических симптомов. К первым относятся: спастический парапарез с повышенными рефлексами, клонусами и патологическими рефлексами Бабинского, Россолимо, Бехтерева, Оппенгейма. К симптомам, симулирующим спинную сухотку («псевдотабес»), относятся парестезии (ощущение ползания мурашек, онемение дистальных отделов конечностей), опоясывающие боли, гипотония и понижение рефлексов вплоть до арефлексии, нарушение вибрационной и глубокой чувствительности, сенсорная атаксия и расстройство функции тазовых органов. У больных могут быть различные психические нарушения, которые варьируют от легкой раздражительности до глубокой деменции и выраженного психоза. Описаны также редкие случаи атрофии зрительного нерва. Необходимо заметить, что выраженность психических нарушений у пожилых больных с дефицитом витамина В12 может иметь самостоятельное, не связанное с анемией значение и хорошо устраняется на фоне терапии витамином В12.
Поражение желудочно-кишечного тракта проявляет себя неприятными ощущениями (жжение) и болями в языке, появлением болезненных трещин в углу рта, снижением аппетита, ахилическими поносами, похуданием. У таких больных может быть выявлен ангулярный стоматит, ярко-красный, с атрофированными сосочками, «лакированный» и болезненный язык, чрезвычайно чувствительный к горячей и кислой пище. Иногда определяется гепато- и спленомегалия. Поскольку большинство этих изменений желудочно-кишечного тракта связано с нарушением синтеза ДНК в слизистом эпителии, они могут в такой же мере наблюдаться и при фолиеводефицитных анемиях.
Что касается анемического синдрома при В12– и фолиеводефицитных анемиях, в нем нет ничего особенного. Как и другие больные анемией, эти больные предъявляют жалобы на общую слабость, утомляемость, головокружение, шум в ушах, сердцебиение, боли в сердце и одышку при физической нагрузке. При осмотре, помимо бледности, может быть обнаружена некоторая желтушность кожи и склер, связанная как с внутрикостномозговым гемолизом плохо дифференцирующихся нормобластов, так и с укорочением продолжительности жизни патологически измененных эритроцитов. Пульс, как правило, частый. Размеры сердца умеренно увеличены. При аускультации на верхушке сердца определяется систолический шум.
Лабораторная диагностика. Диагноз мегалобластных анемий ставится на основании клинического анализа крови, пунктата костного мозга и биохимического определения в крови дефицита витамина В12 и/или фолиевой кислоты. Поскольку патогенетические механизмы нарушения гемопоэза при В12– и фолиеводефицитных анемиях имеют много общего в плане остановки синтеза ДНК, анализы крови и костного мозга при них практически неотличимы.
Так, для обеих анемий характерен макроцитоз, точнее макроовалоцитоз (см. цв. вкл., рис. 5.3), пойкилоцитоз, наличие в крови поломанных клеток, а также эритроцитов с остатками ядер (тельца Жолли и кольца Кэбо). Количество ретикулоцитов снижено. Помимо анемии характерны лейкопения и тромбоцитопения, указывающие на остановку синтеза ДНК и в гранулоцитарном, и в мегакариоцитарном ростках. Прямым отражением этого является наличие в крови гиперсегментированных (более 5 ядерных сегментов) нейтрофилов, а также появление в крови гигантских палочкоядерных нейтрофилов и метамиелоцитов. У многих больных в крови обнаруживаются даже мегалобласты, которые поступают в циркуляцию из очагов экстрамедуллярного кроветворения в печени и селезенке.
Пунктат костного мозга обнаруживает выраженную гиперплазию эритроидного ростка с мегалобластным типом кроветворения и со смещением соотношения между миелоидными и эритроидными клетками в сторону последних. Развивающиеся в условиях остановленного синтеза ДНК мегалобласты обнаруживают отчетливый асинхронизм в созревании ядра и цитоплазмы. В частности, в эритроидных клетках сохраняется нежная структура хроматина, в то время как цитоплазма созревает и накапливает гемоглобин, как обычно. Кроме того, созревающие эритроидные элементы обнаруживают такие необычные черты дисплазии, как многоядерность, микроядра, межъядерные мосты, а также тельца Жолли. Часть метамиелоцитов имеет гигантские размеры. Мегакариоциты, как правило, имеют гиперсегментированные ядра и довольно нежный хроматин.
При биохимическом исследовании может быть выявлено повышенное содержание в сыворотке несвязанного билирубина и лактатдегидрогеназы при нормальном содержании железа и ферритина. Уровень витамина B12 или/и фолиевой кислоты, как правило, снижен. Вспомогательным тестом на наличие в организме дефицита витамина В12 может быть определение в моче содержания метилмалоновой кислоты, которое в случае В12-дефицита будет увеличено.
Дифференциальный диагноз анемий, связанных с нарушением синтеза ДНК, прежде всего следует проводить с апластическими анемиями и миелодиспластическим синдромом. В отличие от апластических анемий, костный мозг больных мегалобластными анемиями богат клеточными элементами и обнаруживает мегалобластное кроветворение. Кроме того, такие анемии, как правило, хорошо поддаются терапии витамином В12 и/или фолиевой кислотой, о чем свидетельствует быстрый подъем ретикулоцитов в крови, что при апластических анемиях не наблюдается. В отличие от макроцитарных рефрактерных анемий, которые представляют собой часть миелодиспластического синдрома (МДС), при В12-и фолиеводефицитных анемиях не обнаруживаются свойственные МДС хромосомные нарушения, например делеция части длинного плеча хромосомы 5.
Лечение больных В12– или фолиеводефицитными анемиями с равным успехом может проводиться и в амбулаторных условиях, и в стационаре. При этом следует иметь в виду, что назначение витаминов при мегалобластных анемиях до получения стернальной пункции неграмотно. Дело в том, что характерное для них мегалобластное кроветворение может исчезнуть уже после одной инъекции витамина В12, что крайне затрудняет и постановку диагноза, и дальнейшее лечение больных.
Для лечения В12-дефицитной анемии можно использовать две лекарственные формы – цианокобаламин и оксикобаламин. При отсутствии клиники фуникулярного миелоза витамин В12 вводится в дозе 250 – 500 мкг/сут, а при наличии такового (чтобы преодолеть гематоэнцефалический барьер) разовая доза увеличивается до 1000 мкг. Первые 7 – 10 дней препарат вводится ежедневно, затем 1 – 2 раза в неделю до полной нормализации гематологических показателей. Одним из ранних признаков эффективности проводимой терапии считается увеличение содержания ретикулоцитов в крови, которые в первую неделю лечения определяются регулярно. При достижении клинико-гематологического эффекта витамин В12, с целью создания депо, вводится дополнительно еще в течение двух недель. Далее, если причиной дефицита витамина В12 у больного был атрофический гастрит или гастрэктомия, ему показана пожизненная поддерживающая терапия витамином B12 в дозе 250 мкг/мес. Если должный эффект при введении витамина B12 отсутствует, базисная терапия может быть дополнена назначением малых доз преднизолона, чтобы устранить нейтрализующее витамин влияние антител, вырабатываемых на его введение.
Для лечения фолиеводефицитной анемии используют фолиевую кислоту в дозе 15 мг/сут, эффективность которой также контролируется повышением ретикулоцитов крови. Сначала она дается до нормализации клинико-гематологических показателей, а затем в течение двух недель для создания необходимого запаса в организме. Побочных эффектов терапии, как правило, нет. Прогноз заболевания хороший.
5.4. АПЛАСТИЧЕСКИЕ АНЕМИИ
Под апластической анемией понимают такое состояние кроветворения, которое характеризуется наличием в крови панцитопении (анемии, лейкопении и тромбоцитопении) при разной степени выраженности гипоплазии или аплазии костного мозга. По природе своей она может быть врожденной или приобретенной, по генезу – миелотоксической или иммунной, а по течению – средней тяжести, тяжелой и сверхтяжелой. Апластические анемии встречаются с частотой 1 – 3,5 случая на 100 тыс. населения.
5.4.1. Врожденная апластическая анемия (Фанкони)
Примером врожденной апластической анемии может быть анемия Фанкони. Она наследуется по аутосомно-рецессивному типу и из-за нарушения синтеза ДНК сопровождается множественными случайными поломками хромосом, эндоредупликацией генома и хроматидными обменами.
Клиническая картина. Болезнь проявляет себя рано. В клинике, наряду с нарушением кроветворения и связанной с ним анемией, может быть разной степени выраженности геморрагический диатез, объяснимый тромбоцитопенией, а также повышенная чувствительность к инфекциям из-за гранулоцитопении. Это проявляется на фоне различных дефектов развития скелета (микроцефалия, отсутствие лучевой кости и больших пальцев кисти, маленький рост), врожденной гипоплазии почек, гипогонадизма, гиперпигментации кожи и т. д.
Наиболее серьезными осложнениями апластической анемии являются инфекционные осложнения и геморрагический диатез, которые сильнее выражены при тяжелой форме болезни, и в первую очередь требуется их коррекция.
Лабораторная диагностика. Анализ крови обнаруживает анемию, тромбоцитопению и гранулоцитопению разной степени выраженности с увеличением относительного содержания лимфоцитов и, возможно, плазматических клеток. Анемия носит явный гипорегенераторный характер с уменьшенным количеством ретикулоцитов.
Пунктат костного мозга скудный и малоклеточный. В трепанатах костномозговые фрагменты бедны кроветворными элементами, и более трех четвертей их замещены жировой тканью. В большинстве участков мало и гранулоцитарных, и эритроидных, и мегакариоцитарных элементов, а доминируют лимфоциты и плазматические клетки. Вместе с тем могут встречаться места нормальной клеточности с выраженными лимфоидными фолликулами.
Диагноз анемии Фанкони предполагается при наличии в клинике отмеченных выше соматических нарушений, которые сочетаются с развитием панцитопении крови. Он подтверждается данными трепанобиопсии костного мозга, а также результатами его цитогенетического исследования, которые выявляют аплазию кроветворения и характерные для анемии Фанкони множественные нарушения хромосом. Решающим для постановки диагноза является анализ хромосом после культивирования клеток с диэпоксибутаном или митомицином С, которые вызывают в этих клетках множественные индуцированные мутации.
Дифференциальный диагноз следует проводить с другими врожденными апластическими анемиями, в частности ассоциирующимися с врожденным дискератозом кожи, нарушением роста волос и ногтей. При этом могут быть еще телеангиэктазии на теле, наблюдаться резкое выпадение волос и нарушение потоотделения. У таких больных может быть и умственная отсталость, но хромосомные нарушения кроветворных элементов для них не характерны.
Лечение врожденной апластической анемии включает трансфузии эритроцитов и тромбоцитов и профилактическое введение десферала или эксиджада. В случае вторичной иммунизации больного на фоне гемотрансфузий могут быть использованы глюкокортикоиды и циклоспорин А. Некоторым больным может быть предложена спленэктомия. Однако радикальным способом лечения анемии Фанкони остается трансплантация костного мозга со всеми вытекающими из нее положительными и отрицательными последствиями.
Прогноз при этом заболевании тяжелый. Приблизительно у 20 % больных болезнь в течение 5 – 10 лет может трансформироваться сначала в МДС, а потом в острый нелимфобластный лейкоз.
5.4.2. Приобретенные апластические анемии
Выделяют две формы приобретенных апластических анемий: миелотоксическую и иммунную.
Этиология. Появлению миелотоксической формы приобретенной апластической анемии способствуют: радиация, цитостатики, инсектициды, мышьяк, органические растворители и ДДТ. В то же время иммунный механизм повреждения костномозгового кроветворения просматривается при действии на клетки таких лекарств, как левомицетин, сульфаниламиды, некоторые уросептики и соли золота. Кроме того, он явно присутствует при возникновении апластической анемии у больных гепатитами (А, В и С), перенесших вирусную инфекцию Эпштейна – Барр, у беременных и т. д.
Патогенез. В основе развития апластических анемий лежит резкое уменьшение в костном мозге количества полипотентных стволовых клеток и неспособность их или стромы обеспечить полноценный гемопоэз. В части случаев это происходит из-за прямого повреждающего действия на стволовые клетки неблагоприятных факторов внешней среды или цитостатиков, в части – через иммунные механизмы. В любом случае недостаточное количество в костном мозге полипотентных стволовых клеток приведет к недопродукции кроветворных элементов всех ростков и уровней созревания, к запустению костного мозга (см. цв. вкл., рис. 5.4) и к развитию панцитопении крови.
Клиническая картина. Клинические проявления апластической анемии могут быть различными. В некоторых случаях она возникает остро и быстро прогрессирует, в других – заболевание течет относительно спокойно. На первое место в клинической картине выступает анемический синдром. Больные жалуются на общую слабость, одышку, сердцебиение и на плохую переносимость физических нагрузок. Углубление тромбоцитопении может привести к подкожным кровоизлияниям, кровоточивости десен, меноррагиям и повторным кровотечениям со стороны слизистых оболочек. Проявлением глубокой нейтропении могут быть рецидивирующие инфекции, включая ангину, стоматит, пневмонию, парапроктит.
В анализе крови выявляется нормохромная анемия разной степени выраженности и сниженное количество ретикулоцитов. Количество лейкоцитов ниже нормы за счет уменьшения числа гранулоцитов. В тяжелых случаях может быть агранулоцитоз, абсолютная моноцитопения и лимфопения. Количество тромбоцитов снижено, в тяжелых случаях до 20 % 109 и ниже.
Диагноз. Мысль об апластической анемии должна возникать у врача при наличии у больного клинических проявлений недостаточности эритроидного (анемия), мегакариоцитарно-тромбоцитарного (геморрагический диатез) и гранулоцитарного (инфекции) ростков кроветворения, а также отмеченных выше изменений крови. Поскольку пунктат костного мозга у многих больных апластическими анемиями получить не удается, адекватным методом диагностики считается трепанобиопсия. Последняя выявляет выраженную аплазию кроветворения с замещением большей части костных фрагментов (> 75 %) жировой тканью. В большинстве анализируемых участков эритроидные, гранулоцитарные и мегакариоцитарные элементы отсутствуют, а встречаются в основном лимфоциты и плазматические клетки. В то же время отдельные фрагменты костного мозга больных апластическими анемиями могут быть достаточно клеточными и содержать лимфоидные фолликулы. При этом считается, что основными критериями для диагноза тяжелой формы апластической анемии являются: количество гранулоцитов менее 0,5 % 109/л; количество тромбоцитов менее 20 % 109/л; скорригированное на нормальные цифры эритроцитов количество ретикулоцитов менее 1 % при клеточности костного мозга менее 30 %.
Дифференциальный диагноз апластической анемии следует проводить с другими видами анемий, в первую очередь с В12– и фолиеводефицитными, миелодиспластическим синдромом, острым лейкозом и метастазами рака в костный мозг. В отличие от апластической анемии, при В12– и фолиеводефицитных анемиях в большинстве случаев миелодиспластического синдрома и острого лейкоза костный мозг богат клеточными элементами. Кроме того, в нем представлены в увеличенном количестве бластные элементы. В случае же аплазии костного мозга из-за метастазирования туда раковых клеток их пласты в трепанатах костного мозга обнаруживаются без большого труда.
Лечение приобретенной апластической анемии представляет собой сложную задачу. Оно начинается с изъятия у больного опасных в отношении аплазии костного мозга лекарств и защиты его от действия других потенциальных индукторов аплазии. По мере необходимости проводится заместительная терапия (трансфузии эритроцитарной массы и тромбоконцентрата). Осуществляется профилактика и лечение инфекционных и грибковых осложнений соответственно антибиотиками, в том числе неадсорбируемыми в кишечнике, и противогрибковыми препаратами (низорал, дифлюкан, нистатин, вифенд, каспофунгин).
Специальные методы лечения тяжелой идиопатической апластической анемии включают: а) антилимфоцитарный глобулин (АЛГ); б) большие дозы глюкокортикоидов; в) циклоспорин-А или сандимун; г) гемопоэтические факторы роста (эритропоэтин, гранулоцитарно-макрофагальный и др.); д) циклофосфан; е) трансплантацию костного мозга.
Назначение АЛГ позволяет получить эффект у 50 – 60 % больных. Чаще всего АЛГ применяют в сочетании с большими дозами метилпреднизолона, который, помимо усиления самой терапии, помогает ослабить такие побочные эффекты антилимфоцитарного глобулина, как лихорадка, гипотензия, сывороточная болезнь и некоторые другие. Возможно также комбинированное лечение тяжелой формы апластической анемии АЛГ, высокими дозами метилпреднизолона и сандимуна, которое, по некоторым статистикам, может быть эффективно у 70 – 80 % больных. Наконец, у некоторых больных с иммунными формами апластической анемии терапию АЛГ можно успешно комбинировать с назначением циклофосфана.
Альтернативным методом лечения апластической анемии может быть пульс-терапия метилпреднизолоном (1,0 г/сут) в течение 3 – 5 дней. Радикальным же методом лечения тяжелой формы апластической анемии являются миелотрансплантации. После трансплантации пятилетняя выживаемость наблюдается у 60 – 70 % больных.
Для лечения апластической анемии средней тяжести применяют глюкокортикоиды, например преднизолон в дозе 1 мг/кг/сут в течение 1 – 2 мес. с последующим постепенным снижением дозы, и андрогены (оксиметалон 2,5 мг/кг/сут). При отсутствии эффекта показана спленэктомия.
5.5. ГЕМОЛИТИЧЕСКИЕ АНЕМИИ
Гемолитические анемии (ГА) представляют крайне разнородную группу заболеваний крови, при которых укорочена продолжительность жизни эритроцитов. По своей природе они могут быть врожденными и приобретенными.
Преждевременное разрушение эритроцитов может происходить двумя путями. Первый, более часто встречающийся, – внесосудистый или внутриклеточный гемолиз, при котором эритроциты разрушаются макрофагами ретикулоэндотелиальной системы. Как правило, он происходит при наличии на поверхности эритроцитов антител, к которым макрофаги проявляют особый интерес.
Другой причиной внутриклеточного гемолиза может быть повышенная врожденная или приобретенная деформабельность эритроцитов, затрудняющая их прохождение через синусоиды селезенки и, отсюда, ухудшающая их выживание. В случае внутрисосудистого гемолиза разрушение эритроцитов происходит непосредственно в сосудистом русле, а их содержимое поступает прямо в кровь. Как правило, повышенное разрушение эритроцитов сопровождается компенсаторным усилением их продукции, нередко в 6 – 8 раз.
5.5.1. Врожденные гемолитические анемии
В эту группу ГА входят анемии с нарушением мембраны эритроцитов (эритроцитопатии), дефектом ферментов (ферментопатии) и дефектом гемоглобина (гемоглобинопатии).
Наследственный микросфероцитоз. Врожденная микросфероцитарная гемолитическая анемия представляет собой группу семейных заболеваний, обусловленных патологией мембраны эритроцитов. У трех четвертей больных заболевание передается по аутосомно-доминантному типу, у четверти – по аутосомно-рецессивному.

Рис. 5.5. Схематическое представление структуры цитоскелета эритроцита человека (Blood. Principles and Practice of Hematology. – Philadelphia: Lippincott, 1995. – Р. 1726): ГФА – гликофорин А; ГФС – гликофорин С
Патология эритроцитов при наследственном микросфероцитозе обусловлена дефектами генов основных мембранных белков цитоскелета эритроцитов (рис. 5.5), а именно спектрина, гены á и â которого расположены в локусах хромосом 1q22-q23 и 14q23-q24.2 соответственно; анкирина (8р11.2 – 70 %), АЕ1 – обменника анионов (17q21-qter – 25 %) или белка 4.2 (15q15). Необходимо отметить, что, несмотря на отмеченные генетические дефекты и аномальные цитоскелетные белки, костный мозг этих больных продуцирует эритроциты нормальной формы. Однако ввиду того что часть мембраны не поддерживается белками цитоскелета, при прохождении синусов селезенки она утрачивается. В результате отношение площади эритроцита к его объему постепенно уменьшается. Клетки становятся сферическими (см. цв. вкл., рис. 5.6), а размеры их отчетливо уменьшаются. Общая деформабельность таких эритроцитов еще больше снижается. В итоге они не могут пройти в отверстия селезеночных синусов, захватываются макрофагами и разрушаются.
Клиническая картина. Клиническая симптоматика наследственного микросфероцитоза включает анемический синдром, желтуху, изменение костей скелета и спленомегалию. Анемический синдром, как правило, выражен умеренно. Проявление желтухи варьирует и во многом зависит от функционального состояния печени. Если она работает хорошо, желтухи может не быть даже при выраженном гемолизе. В случае же функциональной недостаточности печени даже умеренный гемолиз может сопровождаться выраженной желтухой. Почти у всех больных имеет место спленомегалия разной степени выраженности. Чрезвычайно характерны также такие изменения костей, как «башенный череп» и «высокое нёбо», которые являются прямым отражением ненормального развития плоских костей новорожденных в условиях резко активированного гемолизом эритропоэза. Основными осложнениями заболевания могут быть гемолитические и апластические кризы, а также желчнокаменная болезнь.
Диагноз заболевания ставится при наличии у больного повторяющейся в течение жизни желтухи, отмеченных выше изменений со стороны костной системы и спленомегалии при выявлении подобных симптомов у нескольких членов семьи. Подтверждается диагноз обнаружением в крови микросфероцитов (менее 6 μ в диаметре), увеличенным содержанием ретикулоцитов, активацией эритроидного ростка в костном мозге, выявлением укороченной длительности жизни эритроцитов после мечения их Сr51. Осмотическая стойкость эритроцитов при наследственном микросфероцитозе может быть снижена, хотя у некоторых больных это становится очевидным только после суточной инкубации эритроцитов при температуре 37 °C. С другой стороны, спонтанный лизис эритроцитов после 48-часовой их инкубации по сравнению с контролем может быть увеличен во много раз. Содержание гемоглобина крови обычно превышает 90 г/л, но в период кризов может резко снижаться. При обострении может быть отмечено повышение в крови непрямого билирубина, увеличенное содержание в кале стеркобилиногена и стеркобилина, а в моче – уробилиногена и уробилина. При ультразвуковом исследовании печени может быть выявлен холелитиаз.
Дифференциальный диагноз микросфероцитарной анемии следует проводить с другими гемолитическими и негемолитическими анемиями и различными видами желтух, в том числе доброкачественных. В отличие от других гемолитических анемий, диагноз микросфероцитарной анемии ставится с учетом наследственной природы заболевания, возможности характерных изменений плоских костей и выраженности феномена микросфероцитоза.
Отличить микросфероцитарную анемию от негемолитических желтух помогает выраженный микросфероцитоз и ретикулоцитоз в крови, снижение осмотической стойкости эритроцитов, наличие в костном мозге компенсаторной реакции эритроидного ростка на гемолиз, а также обнаружение спленомегалии без признаков портальной гипертензии. В трудных случаях могут помочь молекулярно-биологические исследования генов.
Лечение. Единственным способом лечения микросфероцитарной анемии является спленэктомия.
Наследственный эллиптоцитоз. Другим видом врожденной эритроцитопатии является эллиптоцитоз (см. цв. вкл., рис. 5.7).
В основе заболевания лежат мутации тех же генов á и â цепей спектрина, расположенных соответственно в локусах хромосом 1 (1q22-q23) и 14 (14q23-q24.2), а также белка 4.2 и гликофорина С, гены которых расположены в локусах хромосом 15 (15q15) и 2 (2q14-q21) соответственно. Что касается окончательных причин различия в форме эритроцитов при микросфероцитозе и эллиптоцитозе, они до конца не ясны.
Клиническая картина, принципы диагностики и терапии напоминают таковые при наследственном микросфероцитозе.
Гемолитическая анемия, связанная с дефицитом в эритроцитах глюкозо-6-фосфат-дегидрогеназы. В силу особенностей структуры эритроцита человека, не содержащего ни ядра, ни митохондрий, ни рибосом, единственным источником его энергии является глюкоза. Она диффундирует в эритроцит из плазмы крови и превращается в глюкозо-6-фосфат. Подавляющая часть глюкозо-6-фосфата (до 90 %) используется в ходе анаэробного гликолиза, остальной – с помощью пентозофосфатного шунта. Основным ферментом, участвующим в превращении глюкозы и энергоснабжении эритроцита, является глюкозо-6-фосфат-дегидрогеназа (Г6ФД), которая необходима для синтеза никотинамиддинуклеотидфосфата и глютатиона. В итоге при недостатке данного фермента в эритроците он недополучает и никотинамиддинуклеотидфосфат, и глютатион. В то же время известно, что восстановление глютатиона является основным механизмом защиты гемоглобина и мембран эритроцитов от окисления и, следовательно, от разрушения под действием многих окисляющих веществ, в том числе лекарств.
Дефицит Г6ФД связан с Х-хромосомой и поэтому поражает в основном мужчин. Он зарегистрирован приблизительно у 200 млн человек, живущих в основном в Средиземноморье, на Ближнем Востоке, в Западной Африке и Юго-Западной Азии.
Клиническая картина. Дефицит Г6ФД обнаруживает себя в клинике острым внутрисосудистым гемолизом, который часто возникает на фоне острых инфекционных или каких-то других заболеваний, а также после приема различных медикаментов или еды конских бобов. Клинические проявления гемолиза обычно обнаруживают себя на 2 – 5-й день после воздействия провоцирующего агента. Выраженность симптомов может быть различной – от легкой желтушности до острой почечной недостаточности. Главными же признаками внутрисосудистого гемолиза будут лихорадка (как при малярии), боли в пояснице, появление темной или черной мочи из-за присутствия в ней гемоглобина.
Лабораторная диагностика. В межкризовый период картина крови не изменена. В период криза выявляется анемия, иногда очень тяжелая, с фрагментированными эритроцитами, тельцами Гейнца (окисленный денатурированный гемоглобин) (см. цв. вкл., рис. 5.8) в ретикулоцитах, повышенным ретикулоцитозом. При биохимическом исследовании в крови находят свободный гемоглобин, уменьшение содержания связывающего свободный гемоглобин гаптоглобина и умеренное повышение непрямого билирубина. В моче также обнаруживают свободный гемоглобин, а при умеренном гемолизе и наличии клеточного осадка – гемосидерин. В связи с тем, что количество Г6ФД в ретикулоцитах и молодых эритроцитах выше, чем в зрелых, дефицит этого фермента у больного может быть временно замаскирован. Однако повторное определение Г6ФД после купирования криза выявляет его дефицит.
Диагноз Г6ФД-дефицитной гемолитической анемии ставится на основе характерных особенностей гемолиза. В частности, появления на фоне инфекции или приема лекарств немотивированно высокой температуры тела, болей в пояснице, гемоглобинемии, гемоглобинурии и характерной для нее темной мочи, а также снижения гаптоглобина крови. Окончательный диагноз ставится после выявления у больного дефицита Г6ФД.
Дифференциальный диагноз этой формы гемолитической анемии проводится с пароксизмальной ночной гемоглобинурией (болезнь Маркиафава – Микели) и гемолизиновой формой приобретенной гемолитической анемии (см. ниже).
Лечение показано только при гемолитическом кризе. Оно начинается с немедленной отмены вызвавшего гемолиз препарата (делагил, фенацетин, аспирин в больших дозах, сульфаниламиды, нитрофураны, ПАСК и т. д.). Проводится инфузионное введение физиологического раствора, направленное на профилактику острой почечной недостаточности. В тяжелых случаях проводят обменные переливания крови и методы экстракорпоральной детоксикации. Трансфузия эритроцитарной массы показана только в случае тяжелой анемии. При этом в качестве доноров не должны использоваться родственники больного, у которых также может иметь место дефицит Г6ФД.
Профилактика гемолитических кризов заключается в своевременной диагностике заболевания, в том числе у родственников больного, тщательном опросе больных перед дачей лекарств о наличии гемолитических кризов в анамнезе, а также в объяснении больным необходимости осторожного обращения с лекарствами в будущем.
Гемоглобинопатии – это группа гемолитических анемий, обусловленных врожденным дефектом структуры глобина. Описано около 200 клинически значимых гемоглобинопатий, в основе которых лежат мутации глобина. Однако только некоторые из них сопровождаются развитием анемического синдрома.
Этиопатогенез. В норме кровь взрослых людей содержит три типа гемоглобина. Основным является гемоглобин А (Hb A), состоящий из двух á-цепей и двух â-цепей глобина (α2β2). На него приходится 96 – 98 % всего гемоглобина (см. цв. вкл., рис. 5.9).
В гемоглобинах F и А2, которые составляют соответственно 1 – 2 % или 2 – 3 % от общего содержания гемоглобина, β-цепь глобина заменяет γ-цепь (Hb F – α2γ2) или δ-цепь (Hb A2 – α2δ2). Синтез каждой цепи глобина кодируется отдельными генами, которые находятся на хромосомах 11 и 16. Наиболее часто встречающиеся гемоглобинопатии (S,CиЕ)обусловлены аномалией β-цепей глобина, а α-цепь при этом нормальная. Значительно реже встречается наследование двух различных патологических вариантов β-цепи глобина, по одному от каждого из родителей – двойная гетерозиготная гемоглобинопатия, примером которой является гемоглобинопатия SC. Среди гемоглобинопатий, связанных с Hb S, только гомозиготные или двойные гетерозиготные варианты обнаруживают выраженные клинические проявления. Напротив, нестабильные подварианты гемоглобина встречаются только при гетерозиготных вариантах, поскольку гомозиготные обычно несовместимы с жизнью.
Структурные особенности примерно 90 % аномальных гемоглобинов заключаются в замещении одной аминокислоты. Хорошей иллюстрацией гемоглобинопатии может быть серповидно-клеточная анемия.
Серповидно-клеточная анемия (СКА) – это врожденная гемолитическая анемия, обусловленная носительством гемоглобина с измененной структурой, получившего название Hb S.
Эпидемиология. СКА была описана Herrick в 1910 г. Она встречается во всех регионах мира, хотя чаще в неблагополучной по малярии Африке. В Европе больше всего носителей Hb S приходится на жителей Средиземноморья и Кавказа. С другой стороны, в силу сложившихся исторических особенностей формирования населения в США СКА с 72 000 зарегистрированных больных среди наследственных патологий стоит на первом месте.
Патогенез. Суть заболевания состоит в точечной мутации в 6-м кодоне гена ââ-цепи глобина, следствием чего является замещение глютаминовой кислоты на валин. Включение âs-цепи в тетрамер приводит к образованию Hb S. Нерастворимость деоксигенированного Hb S приводит к его полимеризации и, как следствие, к снижению деформабельности и образованию серповидных эритроцитов (см. цв. вкл., рис. 5.10). В основе заболевания лежит точечная мутация в гене ââ-цепи глобина, расположенного на хромосоме 11 в локусе 11р15.4. В результате точечной GAG D GTG мутации в 6-м кодоне â-цепи глобина формирующаяся в результате трансляции â-цепь глобина получает вместо глутаминовой кислоты валин. В свою очередь, включение таких âs-цепей в состав тетрамера глобина приводит к тому, что из-за плохой растворимости Hb S в условиях деоксигенации или высокой концентрации гемоглобина он полимеризуется. Это приводит к нарушению естественной деформабельности эритроцитов и к их серповидноклеточности (см. цв. вкл., рис. 5.11). Естественно, что, проходя по сосудистому руслу и селезенке, такие эритроциты повреждаются, разрушаются и, более того, ответственны за закупорку части мелких сосудов и ишемию снабжаемых ими органов и тканей.
Клиническая картина. Клинические проявления СКА вариабельны и более выражены при гомозиготном (Hb SS), чем при гетерозиготном (Hb SC) варианте. Обострение заболевания носит кризовый характер. При этом часть больных в межкризовый период чувствуют себя хорошо, в то время как крайне тяжелые варианты могут закончиться летальным исходом еще в раннем детском возрасте. В качестве провоцирующих кризы факторов выступают: 1) инфекции; 2) стресс; 3) обезвоживание и увеличение осмолярности плазмы; 4) снижение парциального давления О2 в крови; 5) холод; 6) применение сосудосуживающих препаратов; 7) некоторые другие реже встречающиеся причины. Как правило, заболевание себя не проявляет до тех пор, пока не произойдет замена Hb F на Hb S, что обычно имеет место через полгода. Ведущее место в клинике занимают периодически возникающие боли в различных участках тела и хроническая гемолитическая анемия, протекающая по типу кризов с внесосудистым компонентом гемолиза. Из других проявлений характерны тяжелые инфекции, как правило, начинающиеся уже в раннем детстве. Кроме гемолитических кризов возможно развитие апластических кризов. Серповидноклеточный криз характеризуется появлением острых болей в костях, спине, животе, конечностях, в области печени и/или селезенки. Как уже говорилось, болевой синдром связан с окклюзией сосудов и развитием инфарктов в тканях, на фоне которых нередко имеет место присоединение органной и полиорганной недостаточности. В частности, та же причина лежит в основе повышенной склонности к инфекциям, когда из-за множественных тромбозов сосудов и синусов селезенки развивается функциональная аспления. Наконец, такой же генез свойственен и неврологическим осложнениям (ишемические и геморрагические инсульты с развитием парезов, параличей, судорог, головных болей, рвоты, фотофобии). Здесь необходимо подчеркнуть, что у больных СКА могут быть немые крупные множественные инфаркты мозга, которые могут приводить к необъяснимому другими причинами снижению интеллекта и памяти.
Крайне характерно, хоть и вариабельно, при СКА поражение паренхимы печени, желчевыводящей системы и почек. Так, желчнокаменная болезнь проявляется уже в детском возрасте. Как вариант серповидно-клеточного криза часто развивается острый печеночный криз с некрозом гепатоцитов, который проявляет себя гепатомегалией, нарастанием желтухи, лихорадкой, увеличением АСТ и билирубина. Как правило, длительность печеночного криза не превышает двух недель, но может индуцировать печеночную недостаточность. Другим вариантом печеночного криза у больных СКА является острый печеночный криз с секвестрацией, который проявляется быстрым увеличением размеров печени и падением уровня гемоглобина в крови. Следующим вариантом печеночного криза может быть острый печеночный криз с внутрипеченочным холестазом, который проявляется выраженной гипербилирубинемией (без увеличения температуры), болями в печени, лейкоцитозом, печеночной недостаточностью и в конечном итоге приводит к смерти. Кроме того, в генезе поражения печени у больных СКА могут участвовать также вирусный гепатит или аутоиммунный гепатит и гемосидероз.
Поражение почечных сосудов и клубочков (см. цв. вкл., рис. 5.10) сопровождается развитием острой или хронической почечной недостаточности. При этом как во время гемолитического криза, так и длительное время после него могут иметь место: гипостенурия, протеинурия, гематурия и снижение клубочковой фильтрации. Неудивительно, что в этих условиях довольно часто развивается вторичная артериальная гипертензия.
Далее больным СКА свойственно поражение сердца и легких, в частности развитие острого инфаркта миокарда или ишемической кардиомиопатии с присоединением тяжелой сердечной недостаточности. В свою очередь, нарушение микроциркуляции в легких и тромбозы легочных сосудов предрасполагают к развитию отека легких и тяжелой легочной гипертензии. Из других проявлений заболевания следует упомянуть о: а) частом снижении зрения, связанном с развитием ретинопатии; б) некрозах и язвенных поражениях кожи; в) возможности изменения конфигурации костей черепа и замедления физического и психического развития пациентов.
Для женщин с СКА характерны дисменорея, поликистоз яичников, фиброматоз молочных желез, а в случае беременности – высокий риск летальных осложнений как для матери, так и для плода, а для мужчин – приапизм, импотенция и, реже, гипогонадизм.
Лабораторные данные. Гипохромные микроцитарные дискоциты в мазках крови свойственны Hb SC, нормохромные эритроциты – Hb SS- и Hb S+ â-талассемиям. При всех вариантах СКА встречается пойкилоцитоз. Характерно также снижение гематокрита, числа эритроцитов и показателей гемоглобина крови как в межкризовый период, так и в период криза. Практически у всех больных имеет место ретикулоцитоз. Он меньше выражен при Hb S+ â-талассемия подварианте, а больше – в период гемолитического криза. Характерно появление Howell-Jolly телец в эритроцитах. В то же время только Hb SS-подварианту заболевания свойственны повышенный лейкоцитоз и тромбоцитоз. Наконец, характерные для СКА увеличение неконъюгированного билирубина, лактатдегидрогеназы, снижение гаптоглобина отражают степень гемолиза.
Диагноз. Мысль о СКА возникает у врача при наличии у больных признаков хронической гемолитической анемии врожденного характера, протекающей на фоне тяжелых инфекций и сопровождающейся ишемическими болями различной локализации. Подтверждает диагноз наличие в мазках крови серповидно-клеточных эритроцитов, а также электрофоретические и молекулярно-биологические исследования, направленные на предмет выявления Hb S. Что касается разграничения различных подвариантов заболевания, в частности Hb SS, Hb SC или Hb S+ â-талассемия, для достижения этой цели необходимо проведение специальных исследований. Среди них: а) электрофорез гемоглобина на целлюлозоацетатной пленке при рН 8,4; б) электрофорез гемоглобина на цитратном агаре при рН 6,2; в) тест на растворимость гемоглобина; г) ДНК-типирование (ПЦР, рестрикционный анализ, аллель-специфическая гибридизация и др.). В меньшей мере разграничению подвариантов СКА помогает анализ морфологии эритроцитов. В частности, серповидные эритроциты имеют место при гомозиготной (Hb SS) СКА, а мишеневидные эритроциты – при сочетании СКА с â-талассемией.
Дифференциальный диагноз следует проводить с другими гемолитическими анемиями наследственного характера, в частности с эритроцитопатиями. В отличие от микросфероцитоза и эллиптоцитоза, в мазках крови больных СКА присутствуют характерные серповидные эритроциты, которые первым двум патологиям не свойственны. Кроме того, для СКА характерны периодически возникающие боли в различных участках тела, нередко заканчивающиеся инфарктами, а также тяжело протекающие инфекции. Вместе с тем для окончательной верификации СКА и ее подвариантов крайне желательно определение гемоглобина Hb S по перечисленным выше методикам.
Лечение. Хотя радикального лечения СКА пока не придумано, основные симптомы заболевания могут быть устранены или смягчены при увеличении объема выпиваемой жидкости, использовании анальгетиков, антибиотиков и, в случае необходимости, заместительной терапии донорскими эритроцитами в объеме, обеспечивающем поддержание гемоглобина крови на уровне 100 г/л. При этом доля Нb S должна составлять < 50 %.
Поскольку увеличение в крови фетального гемоглобина, имеющего более низкое сродство к кислороду, уменьшает выраженность тканевой гипоксии, то назначение препаратов, увеличивающих количество Hb F у больных серповидно-клеточной анемией, можно рассматривать как компонент патогенетической терапии. С этой целью используют: 1) гидроксимочевину (0,15 – 0,3 мг/кг), которая увеличивает содержание Hb F путем торможения рибонуклеотид-редуктазы;
2) 5-азацитидин (гипометилирование ДНК – увеличение экспрессии гена ã-глобина и, отсюда, увеличение Hb F); 3) децитабин (гипометилирование ДНК с увеличением экспрессии гена ã-глобина и нарастанием Hb F); 4) аргинин бутират (ингибитор гистондеацетилазного комплекса); 5) эритропоэтин.
Важным компонентом лечения СКА является профилактика развития гемолитического криза с учетом перечисленных выше факторов риска. В этом плане особое значение имеет профилактика инфекций. В то же время профилактика тромбозов до конца не разработана, поскольку за их генез при СКА ответственны не только гиперкоагуляционный синдром и нарушение реологических свойств крови, но и изменение антитромботической и адгезивной функции эндотелия. По-видимому, более предпочтительно в этой ситуации назначение дезагрегантов, чем прямых антикоагулянтов, хотя в период гемолитического криза может быть оправданно также применение низкомолекулярных гепаринов. Поскольку устранение наблюдающегося во время гемолитического криза болевого синдрома может предотвратить нежелательный для СКА спазм периферических сосудов, уменьшить тканевую гипоксию и дальнейшее нарастание гемолиза, оно патогенетически значимо. С этой целью обычно используют как наркотические, так и ненаркотические анальгетики, вазодилатирующие препараты, антикоагулянты, дезагреганты, гидратацию и ингаляцию кислорода.
При тяжелом течении СКА возможно выполнение аллогенной трансплантации костного мозга, эффективность которой составляет около 70 %.
В качестве профилактики гемосидероза должны применяться хелаторы железа под контролем уровня ферритина. Так как у больных на фоне гемолиза и активного эритропоэза имеет место повышенное потребление фолиевой кислоты, показано профилактическое применение фолиевой кислоты в дозе 2 – 3 мг/сут.
Апластические кризы, которые, как правило, развиваются у детей после парвовирусной инфекции, могут потребовать адекватных трансфузий эритроцитов. В этом случае восстановление гемопоэза обычно происходит спонтанно в течение 10 – 14 сут. Вместе с тем следует помнить, что длительная заместительная терапия донорскими эритроцитами может привести к аллосенсибилизации пациентов, что диктует врачам необходимость применения эритроцитарных сред с учетом индивидуального подбора донора, а также использование фильтрованных, отмытых или размороженных эритроцитов.
5.5.2. Приобретенные гемолитические анемии
Эту довольно обширную группу ГА представляют заболевания как иммунной, так и неиммунной природы. Первые связаны с выработкой в организме агглютинирующих антител различных классов к эритроцитам больного, вторые – с повреждающим действием химических и физических факторов окружающей среды, а также клапанных и сосудистых протезов.
В группу иммунных ГА входят заболевания, при которых разрушение эритроцитов происходит под влиянием антител. Среди них выделяются изоиммунные, трансиммунные, гетероиммунные и аутоиммунные анемии.
В то же время по характеру повреждающих эритроциты антител иммунные гемолитические анемии могут быть: а) с неполными тепловыми агглютининами; б) с тепловыми гемолизинами; в) с полными холодовыми агглютининами; г) с двуфазными гемолизинами, причем появление в крови больного агглютининов способствует развитию внесосудистого гемолиза, а гемолизинов – внутрисосудистого.
Клинический опыт показывает, что наибольший удельный вес в группе приобретенных иммунных ГА (до 97 %) приходится на анемии с неполными тепловыми агглютининами. В большинстве случаев гетероиммунных гемолитических анемий к выработке антител приводят инфекции или лекарства, которые в этом случае выполняют роль провоцирующего синтез антител гаптена. В случае же аутоиммунных ГА ведущее место в патогенезе гемолитического синдрома имеет срыв исходной толерантности лимфоцитов к собственным клеткам организма.
Иммунная гемолитическая анемия с тепловыми агглютининами. Эта форма ГА может начинаться по-разному. Реже имеет место очень бурный процесс, чаще – подострый и даже хронический. В первом случае на фоне полного благополучия у больного неожиданно падает гемоглобин, повышается температура тела и темнеет моча. Причем лихорадка, как правило, тем выше, чем острее протекает сам гемолиз. Селезенка не успевает увеличиться. Костный мозг богат клеточными элементами с расширенным эритроидным ростком. В крови помимо увеличенного количества ретикулоцитов может быть адаптационный лейкоцитоз и выраженный сдвиг лейкоцитарной формулы влево. Нередко встречаются также эритрокариоциты. В других случаях темп развития болезни не столь бурный. Больных больше беспокоят слабость и другие признаки анемического синдрома, хотя может быть зарегистрировано и легкое повышение температуры тела, и увеличение селезенки, и различной степени выраженности желтушность кожи и склер.
Диагноз иммунной ГА с тепловыми агглютининами ставится на основании появления у больного анемического синдрома, не связанного с кровопотерей. Он чаще всего выявляется на фоне какой-нибудь инфекции или приема лекарства, сопровождается разной степени выраженности желтухой, высоким ретикулоцитозом и выраженной адаптационной реакцией эритроидного ростка костного мозга. Гемолиз может быть подтвержден выявлением у больного укорочения жизни эритроцитов и повышением в крови непрямого билирубина. Иммунная природа гемолиза доказывается положительной прямой пробой Кумбса, обнаруживающей наличие фиксированных на эритроцитах больного антител. Если результаты пробы Кумбса будут отрицательны, можно использовать более чувствительный агрегационный тест.
Дифференциальный диагноз иммунной ГА следует проводить с врожденным микросфероцитозом. Поставить окончательный диагноз помогает то, что при микросфероцитозе анемия и связанная с ней желтуха носит отчетливый семейный характер. Она встречается у нескольких членов одной и той же семьи и может сопровождаться отмеченными выше изменениями в костной системе. Кроме того, диагностически значимой в отношении иммунного гемолиза будет положительная проба Кумбса.
Лечение ГА с тепловыми агглютининами начинается с отмены провоцирующих ее лекарств. Неотложная терапия включает назначение глюкокортикоидов из расчета 1 мг/кг массы тела. При отсутствии эффекта начальную дозу преднизолона увеличивают в 1,5 – 2 раза или заменяют его на метилпреднизолон (метипред). При достижении положительного эффекта дозу глюкокортикоидов постепенно снижают. Если на этом фоне наступает рецидив гемолиза, используют ритуксимаб или иммунодепрессанты, в частности азатиоприн (имуран) и сандимун (циклоспорин-А).
Следующей линией обороны считается назначение антилимфоцитарного глобулина и спленэктомия. Положительный эффект операции наблюдается у 65 – 70 % больных, у 15 % урежаются гемолитические кризы, которые теперь проходят без или с уменьшенными дозами стероидов. В то же время у 20 % больных спленэктомия эффекта не приносит. В этих ситуациях могут быть показаны и ритуксимаб, и такие иммунодепрессанты цитостатического действия, как циклофосфан, винкристин (онковин), хлорбутин и альфа-интерферон. В качестве важного стабилизирующего иммунный процесс средства может быть рекомендован также решительный отказ от приема молочных продуктов и животной пищи, которые, как известно, являются мощными естественными аллергенами. К сожалению, несмотря на все эти терапевтические ухищрения, у ряда больных приобретенными иммунными гемолитическими анемиями остановить патологический процесс до конца не удается, что вынуждает больных к длительному приему и глюкокортикоидов, и иммунодепрессантов со всеми вытекающими отсюда последствиями.
Прогноз заболевания достаточно серьезный. Основные осложнения – желчнокаменная болезнь, тесно связанные с терапией глюкокортикоидами различные инфекции, стероидный диабет, язвенная болезнь, артериальная гипертензия, надпочечниковая недостаточность, остеопороз и некоторые другие.
Иммунная гемолитическая анемия с холодовыми агглютининами. Своеобразие этой формы приобретенной гемолитической анемии состоит в том, что она встречается преимущественно у пожилых людей и часто сопровождается резко увеличенной (до 90 мм/ч) СОЭ, которая в термостате «нормализуется». Из других важных клинических и лабораторных признаков должны быть отмечены: выраженная полиагглютинабельность эритроцитов, затрудняющая определение группы крови, наличие в сыворотке крови М-градиента, ложноположительная реакция Вассермана, а также прогрессирующий во времени синдром Рейно.
Диагноз. Мысль о возможности у больного такой необычной формы гемолитической анемии возникает у врача в случае резко увеличенной СОЭ и больших сложностей с определением группы крови. Окончательный диагноз ставится с учетом положительной реакции агглютинабельности донорских эритроцитов I группы в сыворотке крови больного, поставленной в холодильнике, при отсутствии таковой в термостате (37 °C).
Дифференциальный диагноз следует проводить с другими формами гемолитических анемий, в том числе связанных с механическим повреждением эритроцитов. Помогает отличить ее отмеченный выше феномен панагглютинабельности эритроцитов I группы, выраженность агглютинации в холодильнике при отсутствии ее в термостате, а также четкая разница в определении СОЭ при различных температурных режимах.
Лечение. ГА с холодовыми агглютининами на терапию глюкокортикоидами реагирует плохо. Поэтому основной линией обороны считаются иммунодепрессанты, в том числе с цитостатическим действием (имуран, циклофосфан, хлорбутин и мелфалан).
Прогноз заболевания достаточно серьезный. В частности, возможна трансформация в парапротеинемический гемобластоз (множественная миелома).
Иммунная гемолитическая анемия с гемолизинами. При ГА, обусловленной появлением в крови тепловых или, реже, двухфазных гемолизинов, нарушение целостности эритроцитарной мембраны происходит с помощью фиксированного на гемолизине комплемента.
Клиническая картина при ГА с гемолизинами довольно своеобразна. Попадание гемоглобина прямо в кровь, а оттуда в мочу проявляется в клинике повышением температуры тела, болями в пояснице, появлением красной или темной мочи и, редко, присоединением умеренной желтухи. Однако в случае, если свободного гемоглобина в мочу поступает мало или он активно переводится находящимися там клетками в гемосидерин, цвет мочи может быть не изменен.
В анализе крови имеют место анемия разной степени выраженности и ретикулоцитоз. Биохимическое исследование крови в период обострения обнаруживает увеличение свободного гемоглобина и снижение гаптоглобина. Анализ мочи, сделанный в период ее потемнения, обнаруживает присутствие в ней свободного гемоглобина. В то же время при наличии мочевого осадка в анализируемых клетках может быть обнаружен гемосидерин (см. цв. вкл., рис. 5.12). Из других специальных исследований заслуживают внимания проба Кумбса, сахарозный и кислотный тесты, проба на осмотическую стойкость эритроцитов и определение длительности их жизни.
Диагноз ГА с гемолизинами ставится на основании характерной клинической картины и отмеченных выше данных лабораторных исследований.
Дифференциальный диагноз, прежде всего, следует проводить с пароксизмальной ночной гемоглобинурией (болезнь Маркиафавы – Микели) и с другими видами ГА.
В отличие от болезни Маркиафавы – Микели, при которой разрушение патологически измененных и чрезвычайно чувствительных к комплементу эритроцитов происходит в условиях ночного снижения рН крови, при этих видах анемий никакой связи со временем суток нет, положительна проба Кумбса. Помогает постановка сахарозного и кислотного тестов. В основе сахарозного теста лежит способность сахарозы увеличивать связывание комплемента эритроцитами. Для ее постановки берутся 3 пробирки. В первой эритроциты больного смешиваются с сывороткой донора и сахарозой. Во второй пробирке эритроциты донора смешиваются с сывороткой больного и сахарозой. В третьей пробирке эритроциты больного помещаются в собственную сыворотку и сахарозу. При болезни Маркиафавы – Микели максимальный гемолиз будет в первой пробирке, слабее – в третьей и отсутствует – во второй. Объясняется это тем, что при этой патологии резко увеличивается потребление комплемента, и отсюда его содержание будет выше в сыворотке донора, чем самого больного. В то же время неизмененные эритроциты донора в сыворотке больного с пароксизмальной ночной гемоглобинурией не разрушаются. В случае же иммунных анемий гемолиз должен фиксироваться в третьей пробирке, где присутствуют и антитела, и чувствительные к ним эритроциты. В настоящее время выявление клонов пароксизмальной ночной гемоглобинурии достаточно просто осуществляется методом проточной флуорометрии, нацеленной на распознавание дефицитных по анкерному протеину (CD55 и CD59) лимфоцитов.
Что касается разграничения гемолитических анемий с тепловыми и двухфазными гемолизинами, его осуществляют следующим образом. Берут три пробирки с кровью больного. Одну помещают на2чвтермостат, вторую – на такое же время на холод, а третью – на час в термостат и на час – на холод. При наличии в сыворотке крови тепловых гемолизинов гемолиз будет в первой пробирке, а при наличии двухфазных – в третьей. Наконец, гемолиз патологических эритроцитов резко усиливается добавлением к ним слабого раствора соляной кислоты (кислотная проба Хема).
Лечение ГА с тепловыми гемолизинами поначалу не отличается от стандартного. Препаратами выбора являются глюкокортикоиды, доза которых должна быть адекватной, чтобы остановить гемолиз. Положительный эффект терапии оценивается по нормализации температуры. После достижения клинического эффекта, который может быть зарегистрирован у 90 – 95 % больных, доза преднизолона или метипреда постепенно снижается. При этом полная клиническая ремиссия достижима лишь у 5 % больных.
Второй «линией обороны» при гемолитических анемиях с тепловыми гемолизинами является спленэктомия, эффект от которой может быть у 66 – 68 % прооперированных больных. Часть больных после операции снова дают редкие гемолитические кризы, не требующие назначения глюкокортикоидов или купирующиеся ими. У 20 % больных эффекта от спленэктомии достичь не удается, что оправдывает назначение иммунодепрессивной терапии. Согласно накопленному опыту, предпочтительнее использовать циклофосфан или винкристин, чем азатиоприн (имуран), несмотря на то что последний дает значительно меньше осложнений.
5.6. ГЕМОРРАГИЧЕСКИЕ ДИАТЕЗЫ
Геморрагические диатезы представляют собой сложную группу заболеваний, связанных с нарушением сосудистого, тромбоцитарного и/или плазменного звеньев гемостаза. Они встречаются с частотой 10 случаев на 100 тыс. населения. Их лечение требует от врача специальных знаний и большого практического опыта.
механизмом к свертыванию крови является повреждение целостности сосудистой стенки, которое, с одной стороны, активирует адгезию и агрегацию тромбоцитов, с другой (через XII контактный фактор) – запускает внутренний механизм плазменного звена гемостаза (см. цв. вкл., рис. 5.13). Для остановки кровотечения из капилляров и сосудов малого калибра, как правило, достаточно первичной гемостатической пробки из массы адгезированных к коллагену и агрегированных тромбоцитов, в образовании которой принимают также прямое участие фибриноген и фактор Виллебранда (ФВ). Первый обеспечивает контакт тромбоцитов друг с другом, а второй ответствен за контакт тромбоцитов и коллагена.
Основными механизмами остановки кровотечений из более крупных сосудов являются уменьшение их просвета и выпадение фибрина. Фибрин образуется из фибриногена под действием тромбина, который отщепляет от фибриногена препятствующие полимеризации фибринопептиды А и В. В итоге фибриноген превращается в фибрин-мономер. Далее он может трансформировать фибрин-мономерные комплексы, которые под влиянием фибринстабилизирующего фактора XIII превращаются в нерастворимый фибрин. В то же время часть фибрин-мономерных комплексов снова распадается до фибрин-мономеров или поглощается клетками ретикулоэндотелиальной системы.
Классификация геморрагических диатезов:
III. Заболевания сосудов:
1. Наследственные.
2. Приобретенные.
3. Вызванные лекарствами.
III. Нарушения тромбоцитов:
1. Количественные:
а) наследственные;
б) приобретенные.
2. Качественные:
а) наследственные;
б) приобретенные;
в) вызванные лекарствами.
III. Нарушения факторов свертывания крови:
1. Наследственные:
а) один фактор;
б) множественные факторы.
2. Приобретенные: а) один фактор;
б) множественные факторы.
IV. Множественные дефекты системы типа ДВС-синдрома.
Естественными противовесами свертывающей системы крови являются антитромбин III, система протеинов С и S, плазмин и простациклин. Под влиянием гепарина первый способен блокировать практически все основные этапы плазменного звена гемостаза, включая превращение фибриногена в фибрин-мономер. Активированный плазменный протеин С в присутствии протеина S способен расщеплять протеолитически активированные факторы VIII и V и таким образом тормозить образование активного фактора Х и тромбина. Плазмин принимает непосредственное участие в протеолитическом расщеплении нитей фибрина, а при необходимости способен разрушать и другие плазменные факторы свертывания крови. Что касается простациклина, он активно препятствует агрегации тромбоцитов.
5.6.1. Тромбоцитопении
Определение. Под тромбоцитопениями понимаются такие заболевания и синдромы, при которых количество тромбоцитов в крови не достигает 150 % 109/л. Удельный вес тромбоцитопений в группе геморрагических диатезов довольно высок (15 – 20 случаев на 100 тыс. населения). Они могут быть врожденными и приобретенными и вызываются: а) недостаточной продукцией тромбоцитов в костном мозге; б) секвестрацией их в сосудах или в селезенке; в) разведением, в частности, перелитой кровью; г) повышенным разрушением или потреблением тромбоцитов (табл. 5.1).
Таблица 5.1
Основные причины увеличенного потребления или разрушения тромбоцитов

Встречаемость врожденных тромбоцитопений: синдром Алпорта; синдром Бернара – Сулье; врожденный дефицит тромбопоэтина; анемия Фанкони; синдром серых пластинок; аномалия Мея – Хегглина; синдром Вискотта – Олдрича.
Врожденная инфильтрация костного мозга:
а) врожденные лейкемии;
б) врожденный ретикулоэндотелиоз;
в) врожденный мукополисахаридоз;
Врожденная гранулематозная болезнь.
Прием лекарств матерью:
а) этанола;
б) тиазидов;
в) толбутамида;
г) стероидов (эстрогенов или преднизона).
Материнские инфекции:
а) цитомегаловирусная;
б) вируса гепатита;
в) краснухи;
г) оспы.
Иммунные тромбоцитопении (ИТ) представляют сборную группу заболеваний, связанных с выработкой антител к предварительно измененным или даже к неизмененным тромбоцитам. По своей природе они могут быть врожденными и приобретенными, а с учетом механизма образования антител подразделяются на:
а) изоиммунные, или аллоиммунные, при которых разрушение тромбоцитов обусловлено или их несовместимостью по одной из групп крови, или трансфузией реципиенту чужих тромбоцитов при наличии у него ранее выработанных антител к ним;
б) трансиммунные, связанные с переходом к ребенку через плацентарный барьер антител от ранее иммунизированной матери;
в) гетероиммунные, обусловленные первичным нарушением антигенной структуры тромбоцитов вирусом, лекарством или другим агентом, выступающим в роли гаптена;
г) аутоиммунные, при которых из-за срыва толерантности лимфоидной системы лимфоциты начинают вырабатывать антитела к своим исходно неизмененным тромбоцитам.
Как показано в табл. 5.2, важное место среди гетероиммунных тромбоцитопений занимают заболевания, связанные с приемом лекарств. Последние чаще всего выступают в роли гаптена, комплекс которого с иммунными белками плазмы находится на тромбоците, фиксирует комплемент, а затем удаляется из циркуляции макрофагами вместе с несущими иммунные комплексы тромбоцитами. Естественно, что при очередном попадании лекарства в организм сенсибилизированные им больные обнаружат быстрое снижение количества тромбоцитов и, наоборот, их повышение вскоре после отмены препарата.
Таблица 5.2
Лекарства, вызывающие иммунные тромбоцитопении

Аутоиммунная тромбоцитопения представляет собой часто встречающееся заболевание крови. В ее основе лежит образование с последующей фиксацией на тромбоцитах иммуноглобулина G, который и ответственен за их быстрое разрушение в ретикулоэндотелиальной системе.
Недостаток тромбоцитов в крови проявляет себя в нескольких направлениях. Во-первых, не образуется или плохо образуется тромбоцитарная пробка, что приводит к увеличению длительности кровотечения и появлению экхимозов и синяков в местах небольших травм. Во-вторых, из-за недостаточного поступления питательных веществ к эндотелию, главными «кормильцами» которого являются тромбоциты, начинает страдать сосудистая стенка, что, несомненно, способствует и появлению, и прогрессированию геморрагического диатеза.
По течению аутоиммунную тромбоцитопеническую пурпуру можно разделить на острую, рецидивирующую и хроническую формы.
Острая форма болезни более свойственна детям, в равной мере мальчикам и девочкам. Она часто возникает на фоне перенесенных сезонных (зима – весна) вирусных инфекций, в том числе типичных детских инфекций и острых респираторных заболеваний. Промежуток времени между появлением пурпуры и перенесенной инфекцией, как правило, составляет две недели, а средняя продолжительность измеряется 1 – 2 мес. Эта форма благоприятнее, чем хроническая, хотя при отказе от глюкокортикоидной терапии смертельные исходы не считаются редкостью.
Хронической формой преимущественно страдают взрослые, причем женщины в 2 раза чаще мужчин. Нередко заболевание может продолжаться 30 лет и более. Что касается ремиссий, то они в основном заключаются не в нормализации количества тромбоцитов, а в исчезновении геморрагий.
Рецидивирующими формами аутоиммунной тромбоцитопении могут болеть и взрослые, и дети. При этом налицо не только повторные рецидивы заболевания, но и полные ремиссии. В большинстве случаев иммунная тромбоцитопения развивается без отчетливой связи с каким-либо сопутствующим заболеванием. С другой стороны, симптоматические варианты болезни встречаются при хроническом лимфолейкозе, миеломной болезни, хроническом активном гепатите, системной красной волчанке, ревматоидном полиартрите, диффузном токсическом зобе и ряде других заболеваний.
Клиническая картина. Больные обращаются к врачу из-за кровоизлияний в кожу и/или кровотечений из слизистых. Излюбленной локализацией мелкоточечных или петехиальных кровоизлияний является кожа нижних конечностей, где внутрикапиллярное давление выше, чем на верхних. Часть петехиальных кровоизлияний может сливаться, образуя экхимозы. По мере прогрессирования патологического процесса кровоизлияния могут появляться и на туловище, поражать сетчатку глаза и даже мозг. Относительно часто при иммунной тромбоцитопении встречаются носовые кровотечения, гиперполименоррея и кровоточивость десен, особенно после чистки зубов. Характерно также образование синяков в местах инъекций. В то же время кровотечения из желудочно-кишечного тракта, мочевыделительной и дыхательной систем встречаются реже. У части больных иммунными тромбоцитопениями важным клиническим синдромом может быть анемия. В этом случае появляются жалобы на слабость, учащение сердцебиения и головокружение.
Объективное исследование больного выявляет отмеченные выше изменения со стороны кожи и слизистых, может быть анемизация разной степени выраженности. Увеличение селезенки и печени малохарактерно. В то же время у некоторых больных имеет место увеличение лимфатических желез, особенно в области шеи, что в сочетании с субфебрильной температурой тела нередко симулирует системную красную волчанку.
Лабораторная диагностика. Самым простым тестом на тромбоцитопению, который может быть проведен в амбулаторных условиях, является увеличение времени кровотечения. Последнее определяется после нанесения на кожу мелкой раны. Для стандартизации результатов последнюю лучше наносить на средней трети предплечья, в условиях туго наложенной на плечо манжетки от прибора для измерения артериального давления. В норме длительность кровотечения из поврежденного сосуда не должна превышать полутора минут.
Другим важным ориентирующим на тромбоцитопению тестом может быть снижение ретракции кровяного сгустка. Это происходит из-за недостаточного поступления в плазму вырабатываемого тромбоцитами ретрактозима и наглядно проявляет себя невозможностью получения сыворотки из взятой без антикоагулянта свернувшейся крови. В то же время свертываемость крови у таких больных нормальная. Не изменены также каолин-кефалиновый и аутокоагуляционный тесты. Ввиду постоянной циркуляции в крови антитромбоцитарных антител, функциональная активность тромбоцитов больных иммунными тромбоцитопениями может быть изменена, что находит отражение в снижении их способности прилипать к стеклу, а также в нарушении АДФ-, тромбин- и коллаген-агрегации.
Диагноз тромбоцитопении ставится на основании характерной клинической картины и упомянутых выше лабораторных тестов, причем окончательный вывод о наличии у больного такого заболевания делается только после обнаружения в крови уменьшенного (менее 150 % 109/л) количества тромбоцитов. Последние лучше определять вместе с клиническим анализом крови, который сразу же позволит отвергнуть вторичные тромбоцитопении при лейкозах и апластической анемии. Вместе с тем в мазках крови может быть представлено увеличенное содержание мегатромбоцитов. Как следствие повторных геморрагий может развиться гипохромная железодефицитная анемия. Уточнение диагноза требует производства стернальной пункции, которая может обнаружить не только увеличение содержания мегакариоцитов, но и изменение их структуры (гиперсегментация ядра). В качестве вспомогательного диагностического теста может быть использовано определение в плазме и на тромбоцитах больного антитромбоцитарных антител. Если таковые обнаружены, диагноз иммунной тромбоцитопении можно считать доказанным; если нет, то отвергать такой диагноз все-таки преждевременно. Дело в том, что титр указанных антител может быть недостаточным для их идентификации, а наиболее насыщенные антитромбоцитарными антителами пластинки уже захвачены и разрушены макрофагами. Кроме того, следует иметь в виду, что определение антител на поверхности тромбоцитов сопряжено с большими методическими трудностями и поэтому в клинических условиях используется редко.
Дифференциальный диагноз иммунных тромбоцитопений проводят с таковыми неиммунного генеза, а также с симптоматическими тромбоцитопениями. В первом случае помогает поставить диагноз отсутствие у больных иммунными тромбоцитопениями заболеваний сердечно-сосудистой системы, особенно корригируемых постановкой искусственных клапанов сердца или сосудистых протезов, наличие в костном мозге увеличенного количества мегакариоцитов и положительные тесты на антитромбоцитарные антитела. Для исключения лейкозов и аплазий кроветворения учитывают данные клинического анализа крови и соответствующие изменения костного мозга. Для исключения системной красной волчанки, болезни Грейвса и ревматоидного полиартрита учитывают наличие у больных свойственных таким патологиям симптомов и соответствующие лабораторные тесты (LE-клетки, антинуклеарный фактор, антитела к нативной ДНК, ревматоидный фактор, увеличение гормонов щитовидной железы в крови и т. д.).
Лечение. Поскольку некоторые виды ИТ тесно связаны с приемом лекарств или с перенесенной недавно инфекцией, первой задачей лечащего врача является отмена этих препаратов и устранение из организма соответствующего инфекционного агента.
Что касается основных методов специфического лечения ИТ, они следующие:
– глюкокортикоиды;
– иммунодепрессанты (азатиоприн);
– внутривенное введение больших доз IgG;
– антилимфоцитарный глобулин;
– иммунокорректоры (сандиммун);
– спленэктомия;
– малые дозы винкристина или винбластина;
– интерферон-á;
– даназол;
– антитела к СD20 (мабтера);
– ромипластин;
– и другие, реже используемые подходы.
При выборе терапии ИТ учитывают возраст больных. Основным же показанием к ее началу считается наличие у больного выраженного геморрагического синдрома, а не уровень тромбоцитопении.
Начинают лечение с назначения преднизолона в дозе 1 мг/кг веса. Эффект терапии (ослабление или исчезновение геморрагического диатеза, а также увеличение числа тромбоцитов) обычно регистрируется через 5 – 7 дней. При отсутствии эффекта первоначальная доза преднизолона увеличивается в 1,5 – 2 и даже в 3 раза. Показанием для снижения дозы преднизолона (не быстрее, чем 5 мг в 4 дня) будет полная ликвидация геморрагического диатеза на фоне отчетливо наметившейся тенденции к повышению количества тромбоцитов в крови. Если в ходе отмены глюкокортикоидов геморрагический диатез возобновляется или будет зарегистрировано критическое снижение тромбоцитов (ниже 10 % 109/л), терапию преднизолоном заменяют на короткий курс метилпреднизолона (30 мг/кг/сут в течение 5 дней) или подключают к основной терапии азатиоприн (0,05 % 3 раза в день).
Альтернативным подходом к лечению острых форм ИТ является внутривенное (в течение 5 дней) введение больших доз (3 г) хорошо очищенного иммуноглобулина G, который особенно важен у детей и беременных.
Второй «линией обороны» хронической ИТ являются антилимфоцитарный глобулин и/или спленэктомия. Основными показаниями для спленэктомии считаются:
– недостаточная эффективность ранее проводимой терапии глюкокортикоидами, в том числе в комбинации с иммунодепрессантами;
– ограниченные возможности длительной терапии глюкокортикоидами у больных сахарным диабетом, язвенной болезнью, артериальной гипертензией, остеопорозом и некоторыми другими нарушениями обмена;
– способ профилактики будущих катастроф у женщин с ИТ, намеревающихся рожать детей.
Спленэктомия проводится в условиях специализированного хирургического стационара под контролем гемограммы и уровня тромбоцитов. Для предотвращения развития нежелательной надпочечниковой недостаточности плановая доза глюкокортикоидов непосредственно перед операцией должна удвоиться. Кроме того, из-за высокого риска развития пневмококкового сепсиса у спленэктомированных больных все готовящиеся к спленэктомии пациенты должны быть предварительно иммунизированы пневмококковыми вакцинами.
В результате такой тактики ведения больных ИТ стойкого положительного эффекта удается достичь у 90 % детей и приблизительно у 60 – 70 % взрослых. Из других способов лечения, используемых для лечения рефрактерных к стандартной терапии ИТ, заслуживают внимания: а) большие дозы метилпреднизолона; б) иммуносупрессивная терапия циклофосфаном или азатиоприном; в) винкристин, винбластин, колхицин или этопозид; г) низкие дозы антирезус-0 (D) иммуноглобулина G; д) даназол; е) á-интерферон; е) аферез или плазмаферез с антистафилококковым протеином А, которые направлены на удаление из циркуляции антитромбоцитарных антител; ж) большие дозы аскорбиновой кислоты; з) дапсон; и) лечебное анти-СD20-антитело – мабтера; к) ромипластин.
Механизм действия при ИТ алкалоидов барвинка розового (винкристина и винбластина), колхицина и группы алкалоидов подофиллотоксина (вепезид и этопозид) приблизительно одинаков. Он связан с прямым воздействием указанных препаратов на основной опорный белок цитоскелета тромбоцитов и макрофагов – тубулин. Поскольку последний формирует микротрубочки, без которых полноценный фагоцитоз невозможен, эта функция макрофагов во время лечения названными препаратами частично утрачивается, в том числе и в отношении покрытых антителами тромбоцитов.
Обычно винкристин для лечения ИТ вводится внутривенно в дозе 0,5 мг один раз в неделю, и такая терапия продолжается в течение 6 нед. Эффективность терапии оценивают через 2 – 3 нед., а в случае успеха эффект может сохраняться 6 мес. и более. До недавнего времени основным сигналом для остановки терапии винкристином являлась периферическая нейропатия в виде полинейропатии, локальных невритов и парезов, в особенности кишечника, частота которых при использовании вышеупомянутых малых доз винкристина стала минимальной.
Колхицин, подобно винкристину и винбластину, связывается с тубулином и нарушает такие зависимые от микротрубочек процессы в макрофагально-мононуклеарной системе, как фагоцитоз. В результате его применения полные и частичные ремиссии ИТ удается получить у 20 и 40 % больных соответственно.
Что касается этопозида, этот препарат может обеспечить длительные стойкие ремиссии у больных с недавно возникшей ИТ. При этом ожидаемого миелотоксического эффекта от действия этопозида не наблюдалось.
Внутривенные инфузии иммуноглобулина G в дозе 400 мг/кг/сут, вводимые в течение 5 дней, способны поднять уровень тромбоцитов крови выше 50 % 109/л у 83 % леченых пациентов. Одним из объяснений положительного эффекта иммуноглобулина при ИТ является блокада Fc-рецептора макрофагов и отсюда меньшее повреждающее воздействие последних на циркулирующие в крови тромбоциты.
Основными же недостатками терапии большими дозами иммуноглобулина G следует считать ее дороговизну и нестойкость эффекта. Поэтому назначение высоких доз иммуноглобулина G, прежде всего, оправданно для купирования остро возникающего геморрагического синдрома у детей, а также как важный этап подготовки к спленэктомии.
Альтернативным подходом для блокады Fс-рецептора макрофагальномононуклеарной системы у Rh-позитивных больных с ИТ может быть внутривенное или внутримышечное введение анти-Rho иммуноглобулина G в дозах 0,75 – 4,5 мг в течение 1 – 5 дней.
Даназол представляет собой алкилированный в 17-й позиции синтетический андроген с минимальным вирилизирующим эффектом. Клинический опыт показывает, что этот эффект даназола при ИТ во многом зависит от длительности терапии, возраста и пола больных, а также от состояния их селезенки. В частности, женщины реагируют на даназол лучше мужчин. В то же время ранее спленэктомированные больные (50 – 60 %) на даназол практически не реагируют. Иммуномодулирующий эффект даназола при ИТ связывают с уменьшением продукции антитромбоцитарных антител, а также с уменьшением числа Fс-(IgG-) рецепторов на фагоцитирующих тромбоциты клетках. Рекомендуемые начальные дозы даназола должны быть не менее 600 – 800 мг/сут, в то время как для поддержания эффекта достаточно приема 200 мг два раза в неделю.
Положительный эффект á-интерферона при лечении взрослых больных с ИТ связывают с его прямым ингибирующим влиянием на В-клетки. Обычно для получения эффекта он вводится в дозе 3 млн ед. в течение 12 дней. Основные побочные эффекты интерферона – различные аллергические реакции, лихорадка разной степени выраженности, а главное – утяжеление самой тромбоцитопении (из-за его прямого тормозящего влияния на деление костномозговых элементов).
Из других способов терапии ИТ заслуживают внимания: а) ежедневная очистка плазмы от антитромбоцитарных антител; б) удаление антитромбоцитарных антител с помощью плазмафереза со стафилококковым протеином-А; в) использование больших доз аскорбиновой кислоты, вводимых внутривенно; г) применение дапсона; д) внутривенное введение анти-СD20-антитела – мабтеры; е) использование агониста тромбоцитарного рецептора ромипластина. Два последних препарата дороги. Однако положительный опыт использования их при тяжелых, рефрактерных к стандартной терапии формах ИТ, в том числе после аллоТГСК, обнадеживает.
5.6.2. Тромбоцитопатии
Определение. Тромбоцитопатии – это врожденные нарушения тромбоцитарного звена гемостаза, связанные с продукцией костным мозгом неполноценных в отношении адгезии, агрегации или реакции высвобождения (секреции) пластинок. Они наследуются по аутосомно-доминантному или рецессивному типу и имеют однотипную клиническую картину.
Нарушение адгезии тромбоцитов к субэндотелиальным элементам имеет место при болезни Бернара – Сулье. В основе нарушения гемостаза лежит дефект тромбоцитарной мембраны, а именно отсутствие на ней гликопротеина Ib, что приводит к нарушению взаимодействия тромбоцитов с ФВ (см. цв. вкл., рис. 5.14), а через него – с субэндотелиальными структурами сосудистой стенки. Примером заболевания с нарушением фазы первичной агрегации тромбоцитов может быть тромбастения Гланцмана, при которой главную роль в патогенезе нарушений тромбоцитарного звена гемостаза играет дефицит/дефект мембранного гликопротеинового комплекса IIb/IIIa, являющегося рецептором для фибриногена. Взаимодействие последнего с гликопротеином IIb/IIIa и Са2+ необходимо для агрегации тромбоцитов и ретракции кровяного сгустка. В итоге тромбоциты больных тромбастенией Гланцмана не способны реагировать полноценно ни на один из стимуляторов агрегации, в том числе на АДФ, адреналин и коллаген. При этом имеет место также отчетливое снижение ретракции кровяного сгустка.
Клиническая картина. Геморрагические проявления при наследственных тромбоцитопатиях появляются в раннем детстве и имеют однотипный характер. Доминируют петехии и пятнистые кровоизлияния на коже. Они появляются спонтанно или после небольших травм. Характерны также десневые, носовые и маточные кровотечения. Реже встречаются желудочно-кишечные кровотечения и кровотечения во время операций и родов.
Диагноз. Мысль о врожденной тромбоцитопатии возникает у врача при наличии в клинике кровоточивости капиллярного типа, наличии удлиненной кровоточивости на фоне нормального содержания тромбоцитов в крови. Лабораторное обследование позволяет выявить первичное нарушение адгезии, агрегации или секреции в тромбоцитах, которое может сопровождаться также нарушением ретракции кровяного сгустка.
Дифференциальный диагноз тромбоцитопатий следует проводить с другими геморрагическими диатезами, проявляющими себя нарушением функции тромбоцитов, в частности с болезнью Виллебранда. В отличие от тромбастении Гланцмана, при которой нарушены все виды агрегации тромбоцитов, приболезни Виллебранда прежде всего нарушается реакция на ристомицин (ристоцитин), которая корригируется добавлением ФВ. В отличие же от болезни Виллебранда при синдроме Бернара – Сулье добавление ФВ не корригирует выявленный дефект нарушенной агрегации тромбоцитов на ристоцитин, поскольку он связан с отсутствием на тромбоцитах Ib гликопротеинового рецептора для ФВ. Кроме того, при болезни Виллебранда, помимо нарушения в адгезии тромбоцитов, наблюдается снижение коагуляционной активности фактора VIII (VIII: C), поскольку в норме ФВ выполняет роль носителя коагуляционного компонента фактора VIII и защищает его от разрушения в крови. Наконец, при синдроме Бернара – Сулье в мазках крови могут присутствовать гигантские тромбоциты, что для болезни Виллебранда не характерно.
Лечение. Некоторое улучшение функциональной активности тромбоцитов при кровотечении может быть достигнуто с помощью дицинона (внутривенно, per os) или 1 % раствора АТФ (по 1,0 г внутримышечно ежедневно) со жженой магнезией по 0,5 г 3 раза в сутки в течение месяца или внутримышечно 10 инъекций адроксона (по 1 мл 2 – 4 раза в день). Положительный эффект приносит фитотерапия (настои крапивы, тысячелистника и подорожника).
Прогноз при наследственных тромбоцитопатиях, как правило, благоприятный. Естественно, он ухудшается при кровоизлияниях в мозг и в сетчатку глаз, приводящих к церебральным нарушениям и к потере зрения.
5.6.3. Вазопатии
Геморрагические диатезы, связанные с поражением сосудов, могут иметь врожденный и приобретенный характер.
Наследственные геморрагические вазопатии включают болезнь Ослера, синдромы Черногубова – Элерса – Данлоса и Марфана, а также гемангиомы. Их патогенез так или иначе связан с неполноценностью, точнее с неправильным развитием соединительной ткани, в том числе субэндотелия сосудов. Кровоточивость обусловлена малой резистентностью и легкой ранимостью сосудистой стенки, в частности в местах телеангиэктазий, а также слабой стимуляцией в этих участках адгезии и агрегации тромбоцитов.
Кровоточивость при наследственных вазопатиях обнаруживает себя в детском возрасте. Речь идет о легко возникающих геморрагиях в кожу, кровотечениях из слизистых оболочек носа, ротовой полости, желудочно-кишечного тракта. Характерны кровотечения из десен во время чистки зубов. Опасны кровоизлияния в мозг и внутренние органы.
Диагноз ставится на основании рано возникшего геморрагического диатеза у больных с возможными дефектами соединительной ткани, проявляющими себя различными пороками развития, но при отсутствии каких-либо выраженных изменений в тромбоцитарном звене гемостаза. В качестве вспомогательного разграничивающего теста может быть использован аспирин, при назначении которого (0,5 г) у больных с вазопатиями геморрагический диатез должен усиливаться, а при тромбоцитопениях нарастает незначительно.
Лечение наследственных вазопатий носит симптоматический характер. Для остановки кровотечения используют гемостатическую губку, фибриноген, прижигание кровоточащих сосудов и т. д.
Прогноз для жизни относительно благоприятный, хотя оснований для беспокойства у таких пациентов в жизни предостаточно.
Приобретенные вазопатии с проявлениями геморрагического диатеза встречаются в клинике значительно чаще врожденных (табл. 5.3), хотя в клиническом плане мало отличаются от них.
Основные лекарства, вызывающие вазопатии: аспирин, йод; изониазид, мепробамат, метилдопа, пиперазин, хинидин, гипотиазид, мышьяк, толбутамид, аллопуринол, резерпин, индометацин, соли золота, фуросемид, эстрогены, дигоксин, хлорпропамид, хинин, сульфаниламиды, левомицетин, варфарин и др.
Диагноз приобретенных вазопатий ставится при наличии в клинике разной степени выраженности поражений сосудов (атеросклероз, диабетическая ангиопатия, системные васкулиты и др.) в отсутствие отмеченных выше нарушений тромбоцитарного звена гемостаза.
Лечение направлено на коррекцию основного заболевания и неспецифическое устранение симптомов геморрагического диатеза.
Таблица 5.3
Заболевания сосудов, проявляющиеся повышенной кровоточивостью

5.6.4. Коагулопатии
Определение. Коагулопатиями называются геморрагические диатезы, связанные с нарушением плазменного звена гемостаза. Они носят или врожденный (болезнь Виллебранда, гемофилии), или приобретенный характер (ДВС-синдром).
Болезнь Виллебранда является самой распространенной формой среди наследственных геморрагических диатезов. Заболевание передается по аутосомно-доминантному или рецессивному типу и встречается как у мужчин, так и у женщин. Поскольку ФВ необходим для полноценной адгезии и агрегации тромбоцитов и для поддержания антигемофильного глобулина, его недостаток приводит к нарушению как сосудисто-тромбоцитарного, так и плазменного звеньев гемостаза. В свою очередь, наличие двойного дефекта в системе гемостаза приводит к тому, что болезнь Виллебранда, с одной стороны, напоминает тромбоцитопатию, с другой – гемофилию.
Патогенез. Фактор Виллебранда представляет собой гликопротеин с молекулярной массой от 500 до 12 000 – 20 000 кДа (см. цв. вкл., рис. 5.13). Он состоит из однотипных димерных протомеров (субъединиц), которые кодируются геном хромосомы 12, и синтезируется в клеточных структурах эндотелия и мегакариоцитах. Синтезированный в мегакариоцитах ФВ поступает в тромбоциты, хранится в á-гранулах и выбрасывается из последних под влиянием вызывающих агрегацию стимулов. Роль ФВ в гемостазе связана с его мультимерным строением и наличием в каждой из его субъединиц строго дифференцированных доменов связывания с рецептором тромбоцитарной мембраны, коллагеном, фактором VIII и гепариноподобными гликозаминогликанами. В итоге он функционирует как белок – носитель фактора VIII и одновременно опосредует адгезию тромбоцитов к субэндотелиальным структурам поврежденных сосудов. При этом ФВ выполняет роль моста, соединяющего рецепторы тромбоцитарной мембраны Ib и IIb/VIIIa (см. цв. вкл., рис. 5.13 и 5.14), а с другой стороны – коллаген, миофибриллы эластина, гепариноподобный гликозаминогликан. Нарушение путей синтеза и секреции ФВ приводит к возникновению различных форм болезни Виллебранда, различающихся и по тяжести, и по лабораторным проявлениям.
Клиническая картина. Болезнь Виллебранда проявляет себя очень рано. У детей в возрасте от года до 5 лет могут быть подкожные геморрагии и кровотечения из слизистых оболочек полости рта и носа. Причиной повышенной кровоточивости могут быть травмы и различные хирургические вмешательства (тонзиллэктомия, аденоэктомия, экстракции зубов). Наряду с этим бывают и самопроизвольные носовые кровотечения. На фоне вирусных инфекций верхних дыхательных путей последние могут стать профузными и приводить к анемии. При появлении менструаций у девочек они могут продолжаться от недели до месяца и носить характер меноррагий. При тяжелом течении заболевания встречаются спонтанно возникающие желудочно-кишечные кровотечения, апоплексии яичников, кровоизлияния в сетчатку глаза, гемартрозы и даже межмышечные гематомы. Гемартрозы, не столь частые при болезни Виллебранда, обычно поражают коленные, локтевые и голеностопные суставы. При этом клинико-рентгенологическая картина гемартрозов похожа на гемофильные.
В целом клиническое течение болезни отличается большим полиморфизмом клинических симптомов, что в первую очередь связано с различным содержанием и характером структурных изменений ФВ плазмы и тромбоцитов. На основании этих критериев выделяют несколько подвариантов или типов болезни. Первый, наиболее часто встречающийся тип, выделяется на основании уменьшенного содержания в крови структурно неизмененного ФВ и полного набора мультимеров. Тип II может быть охарактеризован нормальным или субнормальным уровнем измененного по структуре плазменного ФВ, в мультимерном наборе которого или отсутствуют высокомолекулярные формы (подтип IIА), или имеет место его повышенная аффинность к Ib-рецептору тромбоцитарной мембраны, что обнаруживает себя ускоренной ристомициновой агрегацией тромбоцитов в плазме больного. Наконец, при самом тяжелом и редко встречаемом типе III ФВ не определяется совсем ни в плазме, ни в тромбоцитах. В течении заболевания отмечаются периоды обострений и ремиссий. При этом в ходе взросления больного тяжесть клинических симптомов ослабевает.
Диагноз. Мысль о болезни Виллебранда возникает у врача при наличии в клинике рано возникающего геморрагического диатеза, обнаруживающего одновременно нарушения и тромбоцитарного, и плазменного звена гемостаза. Диагноз подтверждается следующими лабораторными признаками: 1) увеличение первичной длительности кровотечения; 2) снижение ристомициновой адгезивности и агрегации тромбоцитов, устраняемое добавлением ФВ; 3) удлинение активированного парциального тромбопластинового времени (АПТВ); 4) снижение коагуляционной активности фактора VIII; 5) снижение уровня ФВ в плазме (определяется иммуноферментным методом) или нарушение его мультимерной структуры.
Дифференциальный диагноз болезни Виллебранда прежде всего проводят с гемофилией А. Опыт показывает, что наибольшие диагностические трудности возникают при разграничении легких форм болезней, когда столь характерное для болезни Виллебранда увеличение длительности кровотечения и нарушение адгезии тромбоцитов может быть не представлено. С другой стороны, эти лабораторные признаки могут обнаружиться из-за образования антитромбоцитарных антител на фоне частых переливаний крови у части больных с гемофилиями. В этом случае решающее диагностическое значение будет иметь прямое определение содержания антигена и активности ФВ, которые при гемофилии или не отличаются от нормы, или даже повышены.
Лечение. При легких и умеренных кровотечениях у больных с I типом болезни Виллебранда с нормальным содержанием ФВ в тромбоцитах показано медленное (10 – 20 мин) внутривенное введение аналога естественного антидиуретического гормона (десмопрессина) в дозе 0,3 мкг/кг массы тела в 30 – 50 мл изотонического раствора натрия хлорида. При этом повышение ФВ и антигемофильного глобулина регистрируется уже через 30 мин после начала инфузии и достигает максимума через 1,5 – 2 ч. Он удерживается на таком уровне в течение 12 ч, отчего требует повторного введения. Однако лечить десмопрессином более 3 сут не рекомендуется в связи с опустошением последним гранул хранения ФВ в клетках эндотелия и отсюда – с необходимостью синтеза в них новых порций этого фактора.
Для лечения больных с вариантами заболевания, где ФВ в гранулах хранения отсутствует, используют свежезамороженную плазму или криопреципитат, богатый ФВ. При определении гемостатической дозы препарата руководствуются коррекцией прокоагулянтной активности фактора VIII и первичной длительности кровотечения. Эффективность терапии оценивают клинически – по уменьшению интенсивности и продолжительности кровотечений и рассасыванию кровоизлияний. В случае переливания препаратов крови, содержащих и ФВ, и антигемофильный глобулин, уровень последнего увеличивается пропорционально введенному фактору и поддерживается на хорошем уровне. Необходимость в заместительной терапии антигемофильным глобулином возникает только при профузных и длительных кровотечениях. В этом случае фактор VIII вводится внутривенно до остановки кровотечения каждые 24 ч из расчета 20 ед./кг массы тела. Что касается остановки кровотечений из слизистых оболочек полости рта и носа, для этих целей может быть рекомендовано местное использование и тромбина, и гемостатической губки или марли. Прогноз при различных подвариантах заболевания неодинаков. Он более благоприятен при умеренно выраженной форме болезни Виллебранда. В то же время тяжелое течение болезни, нередко наблюдающееся у больных с III типом болезни Виллебранда, может привести к анкилозу суставов и к тяжелой анемии.
Гемофилии образуют группу наследственно обусловленных заболеваний крови, связанных с нарушением плазменного звена гемостаза. По современным классификациям могут быть выделены гемофилии А, В, С, D и Е.
Гемофилия А – наследственный геморрагический диатез, связанный с нарушенной продукцией антигемофильного глобулина или фактора VIII. Поскольку ответственный за этот белок ген локализован на Х-хромосоме и является рецессивным, гемофилией А в основном болеют мужчины. Женщины же, носители гена гемофилии, имеющие вторую Х-хромосому (гетерозиготы), этим заболеванием не страдают. Исключение составляют лишь девочки-гомозиготы, которые родились в семье больного гемофилией отца и матери – переносчика этой дефективной Х-хромосомы. Кроме того, у 10 – 15 % больных с приобретенной в ходе жизни мутацией гена антигемофильного глобулина гемофилия А может иметь и ненаследственный характер.
Патогенез. Нарушение гена, ответственного за синтез антигемофильного глобулина, чаще всего ассоциируется с продукцией функционально неполноценного фактора VIII (гемофилия А+) или, реже, с его количественным дефектом (гемофилия А–). В любом случае снижение активности фактора VIII приводит к резкому замедлению свертывания крови по внутреннему пути. При этом степень дефицита фактора VIII тесно связана с выраженностью геморрагического диатеза, что находит отражение и в классификации болезни. В частности, при тяжелой форме гемофилии А активность фактора VIII не превышает 5 %, при средней тяжести течения она варьирует от 5 до 10 %, а при легкой – превышает 10 %.
Клиническая картина. Основным симптомом в клинической картине являются периодически повторяющиеся эпизоды кровоточивости. Как правило, они регистрируются в раннем возрасте, но могут появиться и позднее 20 лет. Несмотря на рождение с геном гемофилии, новорожденные дети кровоточат редко, в том числе после перерезки пуповины. До полугода склонность к кровотечениям выявляется у немногих, и они могут появиться при надрыве уздечки языка или подрезании ногтей. Прорезывание молочных зубов проходит без кровоточивости. Однако острые края самих молочных зубов могут быть причиной кровотечений из-за прикусов языка, щек и губ. Когда ребенок начинает вставать и пробует ходить, возможность травматизации резко возрастает. Из-за того, что он падает, появляются гематомы туловища и головы. Когда же ребенок начинает активно ходить и бегать, открываются возможности для кровоизлияния в суставы, прежде всего коленные, локтевые и голеностопные. «Спонтанные» кровотечения (вне травматизации), прежде всего, присущи тяжелым формам гемофилии. С другой стороны, оперативные вмешательства, даже экстракция зубов, обычно сопровождаются повышенной кровоточивостью. Поскольку образование первичной тромбоцитарной пробки при гемофилии не нарушено, эти постоперационные кровотечения у больных гемофилией возникают не сразу, а через несколько часов, но из-за отсутствия выпадения фибрина могут продолжаться днями и даже неделями.